导读:关于金属对金属髋关节假体的相关问题,最近越来越被学者和媒体所关注。尤其是近几个月来,包括Science、BMJ、JBJS、J Arthroplasty等在内的各大学术期刊,英国每日邮报、中国CCTV等大众媒体,以及上个月的AAOS都给予了深入的探讨。
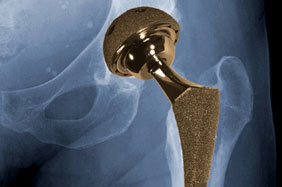
自从上世纪60年代Charnley设计人工髋关节假体并应用于临床以来,人工髋关节置换已经经历了许多重大的改进。但目前人们对最新型的金属对金属髋关节假体顾虑重重,主要缘于这类假体应用于临床后翻修率持续升高,并且诱发肿瘤的风险也明显增加。
文献资料显示,上世纪80年代时全髋关节置换(THA)术后15年内的翻修率低至约20%。当时人们的关注重点在于降低假体无菌性松动和假体周围骨溶解的发生率,这两种现象被认为会缩短假体寿命。THA可以使用多种不同的假体界面材料组合,主要为以下3类:金属对聚乙烯、金属对金属、以及陶瓷对陶瓷。上述名称中前一个名词指的是假体股骨头,后一个指的是臼杯(比如:金属对聚乙烯指的是金属股骨头与聚乙烯臼杯的配对组合)。髋关节表面置换系统则是使用金属材料替代髋关节两侧的表面组织,保留股骨头结构。而传统的THA要求完全切除股骨头,然后将假体股骨头固定于股骨干近端。髋关节表面置换具有术后能更加迅速地恢复髋关节功能并可参与更加活跃的生活方式等优点,因此被认为更适用于相对年轻的患者。但该术式难度相对较高,需要进行专业培训及练习后方能较好地掌握。
2000年,英国国家卫生医疗质量标准署(NICE)发布了一项指导初次THA和髋关节表面置换时如何选择假体的指南,其中针对传统THA设定了术后10年翻修率不高于10%的基准。NICE指出:关节外科医生在术前应该确认拟接受MOM髋关节表面置换的患者已经充分理解以下内容,相对于传统的THA而言,目前对髋关节表面置换器材植入后中长期安全性和可靠性、或术后翻修手术机率等方面的知识仍然知之甚少。
然而,尽管距该指南的发布已经过去了十年,关于这类假体安全性方面的证据仍然十分缺乏。2011年有一篇综述系统性地回顾分析了29篇有关髋关节表面置换的文献,结果发现没有一项研究的结果达到了NICE提出的翻修率不高于10%这一基准值。实际上,2007年一项针对髋关节表面置换的技术评估指出:根据文献资料进行的综述并未跟上髋关节表面置换技术发展的步伐。相关的问题在2008年7月时由于Zimmer公司主动召回旗下的Durom髋臼假体而凸现出来,原因在于该假体临床应用后失败率显著超出预期。随后DePuy 公司于2010年又主动召回其ASR髋关节假体(一种为表面置换系统,另一种为全髋置换系统)。1年后,英国医疗和健康产品管理局(MHRA)向外科医师发出了一封公开信,信中指出:大约40000名接受DePuy公司MOM ASR髋关节假体置换的病人从未收到过产品召回通知。在英国骨科医师协会2011年度年会上,又有更多研究者指出其它公司的大直径MOM髋关节假体同样存在植入后失败率超过预期(在女性患者中尤为明显)的问题。而针对DePuy公司Pinnacle假体存在“金属毒性”的考量更是加剧了人们对MOM髋关节假体的担心。
金属腐蚀现象
制造MOM髋关节假体的材料为钴-铬合金。假体植入体内后部分病人体内这些金属离子水平会明显升高,并可能持续升高,尤其是在那些出现了明显假体磨损以及破坏的病例中更容易出现这一问题。这些金属离子对身体的潜在危害已经非常清楚,有关职业暴露也已有了相应的限制标准。工作场所化合物质对健康危害调查委员会(MAK委员会)对其制定的相关金属离子水平标准值通常会根据最新的研究结果随时更新。2006年,英国按照工作场所金属离子暴露情况制定的钴和铬离子的致癌物质暴露当量(EKA值)分别为:全血中钴离子浓度为5.0μg/L,红细胞中铬离子浓度为17μg/L。而在一项426例接受ASR假体置换病例的研究中发现,总共有28%的病例(女性患者中的比例为41%)体内钴离子浓度超过5.0μg/L。另一项研究发现,111例伯明翰(BHR)髋关节假体植入后仅仅2年时间,体内铬离子和钴离子平均值就已经高达5.1μg/L和4.3μg/L。
对金属假体植入后体内钴、铬离子浓度升高可能导致风险的担心最早始于40年以前。大直径MOM界面的使用必然导致机体各系统在更大程度上面临钴离子和铬离子暴露的风险。有研究发现翻修术时于假体邻近组织所获得的血液和骨髓细胞标本中存在更大程度的染色体畸变率。
尽管相当多的群组研究并没有发现THA术后发生肿瘤的风险增高,但最近一项回顾性研究在探讨接受MOM髋关节置换病例的死亡原因时发现:与传统的金属对聚乙烯THA相比,MOM置换术后第一个20年内因肿瘤死亡的比例更高,但20年后则无明显差别。还有研究发现,术后不同随访期限内有短暂的造血细胞性肿瘤发生率增加,同时前列腺癌和恶性黑色素瘤的发生率也有少许升高。然而大部分随访研究的时间都比较短,而短期内金属离子的浓度通常都会有有较大程度的变化,这提示需要进行长期持续性的观察。同样对于接受髋关节置换的病人也需要确定一个安全的体内金属离子水平。
同样早在40年前就人们就已经注意到钴离子毒性导致的不良反应,包括:恶心、呕吐、神经损伤、以及甲状腺和心血管系统病变。针对工人的流行病学研究已经发现吸入铬离子会导致肺癌发生的风险增高。铬离子同样会对生殖系统造成影响,美国食品和药品管理局(FDA)已经指出MOM假体不宜植入育龄妇女。研究发现接受这类髋关节假体置换后的妇女所生的新生儿体内钴离子浓度有明显升高。
假体设计的缺陷
研究发现,ASR髋关节假体置换后6年内失败率可能高达50%。其主要原因有两个:第一来自于ASR假体设计本身,即其臼杯深度太浅。MOM假体界面工作的原理在于通过髋关节本身产生的关节液润滑金属界面而重建髋关节的运动。当臼杯深度过浅时,假体股骨头在活动中碰撞到臼杯边缘的趋势将显著增加,这将使得假体界面润滑不足而显著加速假体的磨损。随着磨损颗粒的产生,髋关节中将积聚高浓度的铬离子和钴离子,通过一系列累积反应导致广泛的软组织坏死和骨组织破坏。随后,金属离子将扩散进入血液循环并通过肾脏排出体外。
第二个原因是,ASR假体很不幸地同时具有其它MOM大直径假体普遍具有的问题:假体股骨头植入体内后会在其与股骨柄的连接部(即锥度连接部)产生过大的应力,从而释放出磨损颗粒。过去十年在假体设计中对股骨柄作出的某些改变可能与此有关。
监管漏洞
目前,THA或髋关节表面置换已经是临床上常见的手术操作,但MOM假体在临床使用中出现的一系列问题显示相关管理者、假体生产企业、以及医疗机构等相关部门并未就处理好相关问题并预防更多问题的发生作好准备。
欧洲委员会的解决办法是强制所有的欧盟国家使用欧洲中央数据库所认可的医疗器械(Eudamed)。其目的在于便于监管者能尽快地取得相关信息,从而加强医疗器械获得批准后在临床使用中的监管。但该数据库并不对公众,包括研究者开放,而且是由欧盟内部各个国家的相关专家而不是假体生产厂家提供相关数据。这样做会导致相当多的问题,并且这样做是否有助于提高安全性具有很明显的不确定性。
但是最大的一个问题存在于新型器材的认证管理过程中。欧盟在对医疗器械生产厂商提出的规范中指出:临床评估的深度及广度应该是具有一定弹性的,其操作难度不能过大。考虑到一些医疗器械所具有的潜在风险,这种提法让人感到十分奇怪。这还不是唯一让人担心的地方,目前的CE认证监管机制允许仅仅基于已经存在的技术,而不是实际的器械来进行髋关节置换的临床评估。同时,上报的临床数据也仅仅是通过复习文献所获得的。直到2007年2月,髋关节置换假体还被认定是第二类医疗器械,此后才将其归类为与其它植入性器械,比如除颤器一样具有最高风险的第三类器械。第二类器械的临床应用无需提供更多的相关证据,无需对病人的安全性进行评估。实际上,直到2010年随着过度期的结束,才要求市场上所有的髋关节假体必须符合更加严格的标准。
即使假体材料通过了认证,临床应用中也不断有失败病例出现。MHRA早在2008年就已经收到有关ASR假体危及人体健康的证据,然而仍然在过了长达两年的时间之后MHRA才发布了针对医疗器械的警告(MDA/2010/033),要求针对MOM置换患者进行系统性的随访观察,包括在适当的时候检测血液中金属离子浓度,以及对相应关节进行断层扫描检查。而且MHRA在一定程度上弱化了相关问题的严重性,它认为:绝大多数接受MOM髋关节置换的病人术后髋关节功能良好,发生严重问题的风险很低。在此期间,MHRA使用的是来自于英格兰和威尔士国家关节登记库(NJR)的相关信息来指导其作出相关决定。
但问题在于NJR的信息主要反映的是髋关节翻修情况。这需要患者已经出现相关症状并到医师处就诊,关节外科医师通过评估其相关资料确定是否需要进行翻修手术处理。只有对关节进行翻修处理后,相关信息才会进入到NJR的数据库中。随后还要再过相当长的一段时间这些信息才能得到系统分析并公布出来。而且实际上对于一些病例“翻修”的定义是模糊不清的。
在美国也存在类似的问题。美国的多数髋关节假体是按照FDA510(k)方案来进行评估的,产品制造商只需要证明其产品与其它已经投放市场的器材“基本上相似”就可以了。2010年12月31日,FDA依据该方案取缔了包括DePuy ASR假体在内的175项入市申请。目前其它许多产品也已经开始出现失败。
美国医学会最近提出,FDA现行的医疗器械认证体系是不能达到其监管目的的,并建议取消510(k)程序。作为回应,美国参议院提出了一项法案,允许FDA强制产品制造商在其产品通过认证后必须跟踪了解产品的临床使用表现,同时要求加强对产品召回的监管。但与此同时,产品制造商又在游说相关部门,希望能允许其产品尽快地投放市场。医学会的报告指出:FDA应该更好地将其有限的资源投放到制定出完善的产品入市前后的监管体系上面来。
没有哪项产品入市前的管理体系能保证所有的医疗产品都是安全的,但相关部门显然能够做到让投放到市场的器械尽可能安全。目前至少应该努力消除使用包括510(k)程序和CE认证体系在内的多种方案进行监管的混乱局面。针对植入性器械进行入市后的评估分析、提高围绕对于医疗器械警告及退市政策的国际合作等需要制定出一套独立的系统来理顺当前的混乱局面。






