实验目的:
握蛋白质电泳检测常用技术:SDS-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamide gel electrophoresis,SDS-PAGE),以及聚丙烯酰胺凝胶的考马斯亮蓝(Coomassie Brilliant Blue,R-250)染色技术。同时此实验还要学生掌握如何检测分析蛋白原核表达的状态,即判断蛋白原核表达部位:培养液(medium)、可溶性胞质部分(soluble fraction)、周质部分(periplasmic fraction)、包涵体部分(insoluble fraction)。
实验原理:
聚丙烯酰胺(polyacrylamide,PAA)凝胶是丙烯酰胺(acrylamide,Acr),在交联剂甲叉双丙烯酰胺(bisacrylamide,Bis)的作用下经聚合而形成的一种大分子化合物(图12-1)。
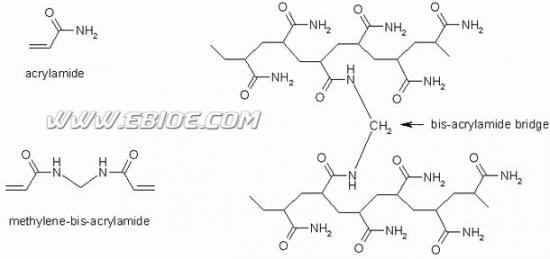
图12-1
丙烯酰胺的聚合作用只有在自由基存在时才能发生,需要一个催化诱发剂系统产生自由基,过硫酸铵(ammonium persulfate,APS)和N,N,N′,N′-四甲基乙二胺(tetramethylethylenediamine,TEMED)就是这样的催化诱发系统。聚丙烯酰胺的聚合反应是通过加入过硫酸铵启动的,添加TEMED加速聚合反应。过硫酸铵提供引发丙烯酰胺和双丙烯酰胺聚合的自由基。诱发剂TEMED能加速过硫酸铵形成自由基,使丙烯酰胺单体转变为自由基态。被激活的单体和未被激活的单体开始了多聚链的延伸,正在延伸的多聚链也可以随机地接上双丙烯酰胺,使多聚链交叉互连成为网状立体结构,最终多聚链聚合成凝胶状。溶液的pH对聚合作用是重要的,因为过低pH没有足够的碱基加速催化反应,同样过多的氧分子存在,会使聚合作用很快停止。所以制备凝胶时,在加过硫酸胺之前,混合物必须抽去空气。
在SDS-聚丙烯酰胺凝胶电泳中,由于SDS和巯基乙醇作用,使得蛋白质分子中二硫键还原,氨键、疏水键打开,SDS按l.4g SDS/1g蛋白质的比例结合到蛋白质多肽链分子上,形成SDS-多肽复合物。因十二烷基磺酸根带负电,使各种SDS-多肽复合物都带有相同密度的负电荷。
它的量大大超过了蛋白质多肽分子原有的电荷量,因而掩盖了不同种类蛋白质多肽分子问原有的电荷差别。同时,在水溶液中,SDS-多肽复合物具有近似的形状(长椭圆棒状),并且不同SDS-多肽复合物的短轴长度趋于一致,而长轴与分子量成正比(图12-2)。这样。SDS-多肽复合物,在SDS-聚丙烯酰胺凝胶电泳的迁移率,不再受蛋白质多肽原有电荷和形状的影响,而只与其分子量有关,并且在一定条件下与迁移率成线性关系。
蛋白质的聚丙烯酰胺凝胶电泳过程中有三种物理效应:①样品的浓缩效应;②凝胶的分子筛效应;③一般电泳分离的电荷效应。因此,样品分离效果好,分辨率高。
SDS-聚丙烯酰胺凝胶的有效分离范围取决于凝胶中聚丙烯酸胺的浓度和交链数量。在没有交链剂的情况下,丙烯酰胺的聚合反应形成黏性溶液,这种溶液是没有实际应用价值的。双丙烯酰胺形成的交链增加了凝胶的硬度和韧性,形成了SDS-多肽混合物通过的孔。这些孔的大小随着双丙烯酰胺:丙烯酸胺比例的增加而变小,当这个比例达到约1∶20时这些孔的孔径达到最小。大多数SDS-聚丙烯酰胺凝胶是由一摩尔的双丙烯酰胺与29 摩尔的丙烯酰胺所组成,这是一个能够按大小分辨多肽而误差小于3%的经验比例。这样,凝胶的过滤性质是由孔的大小而决定的,而这些孔的大小是由加人到凝胶中的丙烯酰胺和双丙烯酰胺的绝对浓度所决定的。凝胶聚合时所形成的微孔影响了不同SDS-多肽复合物电泳时的迁移率,即所谓的分子筛效应(图12-3)。微孔的大小由于Bis和Acr的比例和凝胶的浓度决定。
表12-1列出了凝胶浓度与有效分离范围。
图12-4显示了不同浓度聚丙烯酰胺凝胶对同一样品中各蛋白组分的分离效果。
表12-1蛋白质在聚丙烯酰胺的有效分离范围
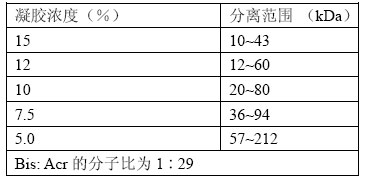
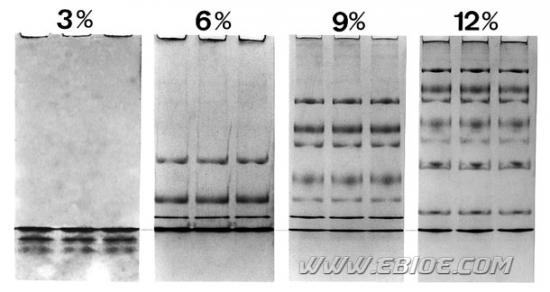
图12-4 不同浓度SDS-聚丙烯酰胺凝胶电泳分离同一蛋白样品的结果
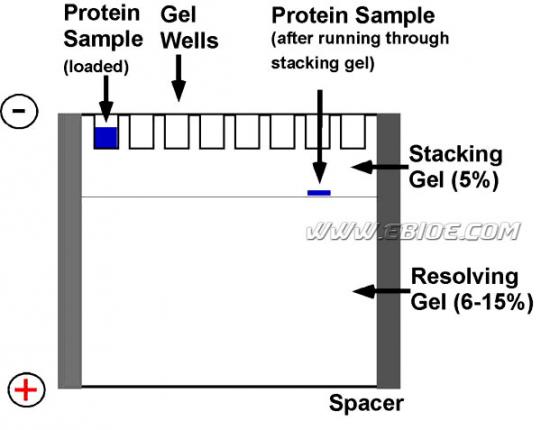
图12-5 浓缩胶和分离胶、上样及样品在浓缩胶中的聚集
多数情况下,SDS-聚丙烯酰胺凝胶电泳使用一种不连续的缓冲液系统。在这一系统中,一般凝胶分为低浓度的浓缩胶(也称“堆积胶或成层胶”)和较高浓度的分离胶 (图12-5、12-6)。配制凝胶的缓冲液也有不同的pH和离子强度。在聚丙烯酰胺凝胶电泳过程中,在移动慢的拖尾离子(甘氨酸)和移动快的前导离子(氯离子)之间形成了一个界面,这是一个通常根据折射系数不同而形成的可见的窄窄的条带。移动界面的两边界之间是一电导较低而电位梯度较陡的区域,推动样品中的多肽前移并在分离胶前沿积聚。当蛋白质样品在浓缩胶中电泳时,蛋白质富集在前导离子和拖尾离子之间的一个窄窄的条带中,SDS-多肽复合物沿移动的界面迁移,最终在分离胶表面聚集成一条很细的区带(积层),极大地浓缩了样品的体积,这就是所谓的浓缩效应(图12-5)。分离胶pH较高,有利于甘氨酸的解离,形成甘氨酸离子穿过堆积的多肽并紧随氯离子之后,在分离胶中泳动。SDS-多肽复合物从移动界面中解脱开来,在电位和pH均匀的分离胶区段泳动,通过分离胶的分子筛作用按分子大小不同得到分离(图12-6、12-7、12-8)
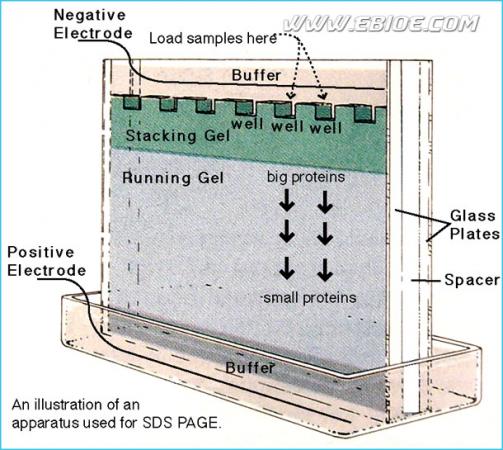
图12-6 垂直电泳装置及不连续胶的位置、蛋白上样及电泳方向
SDS-聚丙烯酰胺凝胶中分离的蛋白质可通过考马斯亮蓝染色(Coomassie staining)或银染色(silver staining)在胶上直接显示观察。一块胶可先做考马斯亮蓝染色,之后脱色再进行银染色。两种染色程序都和蛋白质的质谱分析兼容。或者,蛋白质可以通过电转印(electroblotting)转移到聚偏二氟乙烯(PVDF)膜上,进行Western blot分析(实验13)。
考马斯亮蓝是一种氨基三苯甲烷染料,可与蛋白质形成较强的非共价复合物,复合物的形成可能是范德华力及NH3+基团的静电作用共同作用的结果。考马斯亮蓝用于聚丙烯酰胺凝胶电泳后的蛋白质染色,蛋白质对染料的吸附量与蛋白质的量大致成正比,符合比尔-朗勃定律。
目前已经建立了很多用银盐对SDS-PAGE分离的多肽染色的方法,基本原理都是利用银离子结合在氨基酸侧链上之后还原程度的差异。常用的银盐有银铵溶液和硝酸银,检测灵敏度都高出考马斯亮蓝R-250染色100~1000倍,且能在一条带上检测出0.1~1.0ng的多肽。因硝酸银溶液更容易配制,且不会产生像银铵盐染色中产生的易爆副产物,实验中应用得更多。
实验材料:
[1].30%聚丙烯酰胺单体贮液: 14.55 g丙稀酰胺 + 0.45 g N,N-甲叉双丙稀酰胺,先用40ml双蒸水溶解,搅拌,直到溶液变成透明,再用双蒸水稀释至50 ml,0.45μm滤膜过滤。用棕色瓶储存于 4℃。
[2].分离胶缓冲液:1.5M Tris-HCl, pH8.8。 配制方法:9.08 g Tris溶解在40ml 双蒸水中,用4 mol/L盐酸调节pH值8.8,再用双蒸水加至50ml。4℃保存。
[3].浓缩胶缓冲液:1.0M Tris-HCl, pH6.8。 配制方法:6.06 g Tris溶解在40 ml双蒸水中,用4 mol/L盐酸调节pH值6.8,再用双蒸水加至50 ml。4℃保存。
[4].10 % SDS(十二烷基硫酸钠)(电泳级):10g SDS,用双蒸水溶解并定容至100ml,室温保存。
[5].10% 过硫酸铵:0.1 g过硫酸铵溶解于 1ml双蒸水中。过硫酸铵会缓慢分解,应每周新鲜配制。
[6].TEMED(N,N,N′,N′-四甲基乙二胺): 原液使用。应使用电泳级TEMED。
[7].5×Tris-甘氨酸电泳缓冲液:125mM Tris,1.25M甘氨酸,0.1% (m/v) SDS,pH8.3。配制方法: 在900 ml去离子水中溶解15.1g Tris碱和94 g甘氨酸(电泳级)(pH8.0),然后加入50 ml 10% SDS(电泳级),用去离子水补至1000 ml。
[8].Tris-甘氨酸电泳缓冲液:25mM tris,250mM甘氨酸,0.1% (m/v) SDS,pH8.3。
[9].1×SDS样品缓冲液:50mM Tris-Cl (pH6.8),100mM DTT, 2% (m/v) SDS,0.1%(m/v)溴酚蓝,10%(v/v)甘油。不含DTT的1×SDS样品缓冲液可在室温保存。DTT配成1M 的贮存液,临用前加入。
[10].2×SDS上样缓冲液:100mM Tris-HCl (pH6.8),200mM DTT,4% (m/v) SDS,0.2% 溴酚蓝,20%甘油。
[11].5×SDS样品缓冲液:250mM Tris-HCl (pH6.8),500mM DTT,10% (m/v) SDS,0.5%(m/v) 溴酚蓝,50%(v/v)甘油。不含DTT的5×SDS样品缓冲液可在室温保存。DTT配成1M 的贮存液,临用前加入。
[12].蛋白质分子量标准物(marker)
[13].考马斯亮蓝凝胶染色液:将0.25 g考马斯亮蓝(Coomassie Brilliant Blue)R-250溶解在90ml甲醇:水(1:1,v/v)和10ml冰乙酸中,用滤纸过滤,室温保存。
[14].20% 硝酸银(silver nitrate,AgNO3)溶液:用去离子水配制,棕色瓶室温避光保存。也可以使用前直接配置:0.625g硝酸银溶解于250ml去离子水中。
[15].1M IPTG(异丙基硫代-β-D半乳糖苷):在8ml蒸馏水中溶解2.383g IPTG后,用蒸馏水定容至10ml,用0.22μm滤器过滤除菌,分装成1ml小份贮存-20℃。
[16].10% 三氯乙酸(TCA):50g TCA中加入22.7ml水,形成的溶液含有10%(w/v)TCA。
[17].细胞裂解液:1mg/ml 溶菌酶(lysozyme), 20% (m/v) 蔗糖(sucrose), 30 mM Tris-HCl, 1mM EDTA, pH8.0。
实验步骤:
1、准备SDS-PAGE凝胶
[1].按产品说明将制胶玻璃板装配到制胶器上。
[2].装配好后,可加水检查是否密封,防止漏胶。
[3].确定凝胶浓度体积,按表12-2配制分离胶溶液。按表中成分顺序加入烧杯配制。
[4].小心将分离胶注入准备好的玻璃板间隙中,灌到足够高度。
注意:为浓缩胶留有足够空间(分离胶面距加样孔底1cm,即梳齿长再加1 cm。)。
[5].轻轻在顶层加入几毫升覆盖物(无水乙醇、去离子水或正丁醇),以阻止空气氧对凝胶聚合的抑制作用。
[6].凝胶充分聚合。时间约30~60min, 聚合后在覆盖层和凝胶的界面间有一清晰的折光线。
[7].倒掉覆盖层,用无离子水洗凝胶上部数次,尽可能用吸水纸吸干凝胶顶端的残存的液体。
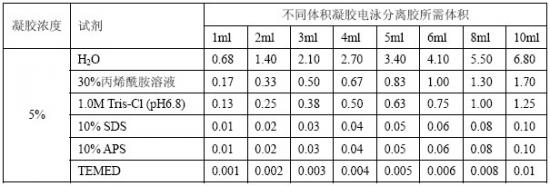
[8].按表12-3配制浓缩胶,并注入分离胶上端。插入梳子。插入梳子子应小心避免梳子顶端留有气泡。
表12-2 Tris-甘氨酸SDS-PAGE分离胶配方
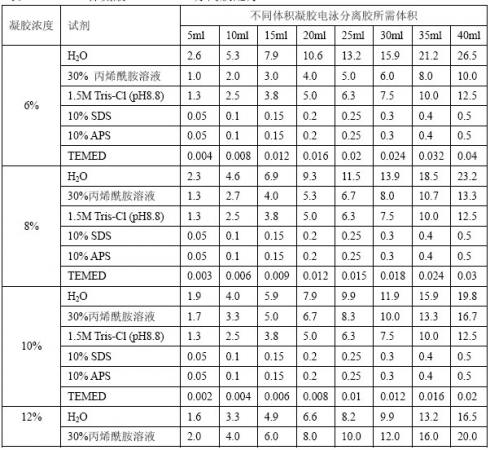
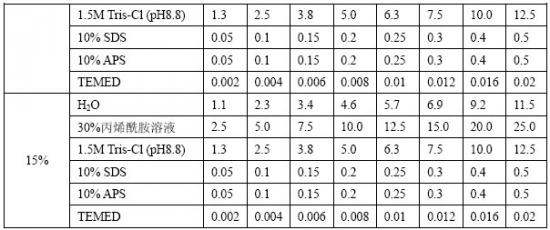
表12-3 Tris甘氨酸SDS-PAGE浓缩胶配方
[9].浓缩胶充分聚合,时间30~60min。
[10].浓缩胶聚合好后,将制胶装置从基座上取下,放如电泳槽。
[11].上下槽均加入新鲜Tris-甘氨酸电泳缓冲液(至少负极槽使用新鲜电泳缓冲液)。
[12].小心拔出梳子,若孔间隔离凝胶条倾斜,用小号注射器针头扶正。
[13].用电泳缓冲液冲洗梳孔。
2、按下列步骤实验,确定蛋白表达与否:
[1].挑取一个单克隆菌落,转入含氨苄青酶素(60μg/ml)的LB培养液,37℃振摇培养过夜;
[2].取适量菌液,按1:100放大培养,A600达到0.4~0.5时,加入0.1mM IPTG继续培养5h,离心收集细菌。
[3].弃去上清,细菌沉淀中加入100μl 0.1M Tris-HCl (pH8.0),重悬细菌沉淀。
[4].加入100μl 2×SDS上样缓冲液,100℃煮沸10min,4℃冷却。
[5].浓缩胶聚合期间,准备样品:将样品与SDS样品缓冲液混合,使SDS样品缓冲液终浓度为1×,100℃加热3~5min。同样准备蛋白质分子量标准物。
注意:如样品是蛋白质沉淀物(如沉淀的蛋白质、细胞、细菌等),加入50~100μl 1×SDS样品缓冲液溶解,并在100℃加热3~5min;若蛋白质为稀溶液,可考虑使用5×SDS样品缓冲液,以增加蛋白质的加样量。蛋白浓度高时,可用2×SDS样品缓冲液。含蛋白质样品的SDS样品缓冲液,如未经100℃加热灭活蛋白酶,切勿放于室温。
[6].取15μl蛋白样品上样,12% SDS-PAGE电泳,电极缓冲液为Tris-甘氨酸电泳缓冲液。
[7].按次序上样,一定安排已知分子量的标准物。
注意:如有空置的加样孔,须加等体积的空白1×SDS样品缓冲液,以防相邻泳道样品的扩散。注意加样时间要尽量短,以免样品扩散,为避免边缘效应。
[8].电泳。开始时电压为8V/cm凝胶,染料进入分离胶后,将电压增加到15V/cm凝胶,继续电泳,直到溴芬蓝染料带抵达分离胶底部1cm距离,断开电源。
[9].取下凝胶,考马斯亮蓝或银染色染色观察,或进行western blot等实验。
[10].按下面的方法对凝胶进行考玛斯亮蓝染色或银染分析。
2、制备不同细菌细胞部位的蛋白样品,确定蛋白表达的部位:
[1].挑取一个单克隆菌落,转入含氨苄青酶素(100μg/ml)的LB培养液,37℃振摇培养过夜;
[2].取适量菌液,按1:100放大培养,A600达到0.4~0.5时,加入0.1mM IPTG继续培养5h,离心收集细菌。
[3].转移上清,上清液中加入等体积冰浴冷的10% TCA,冰上放置20min,高速离心后弃上清,用1ml甲醇洗涤沉淀2次,标记为A,即菌液部分。
[4].在细菌沉淀中加入100μl 细胞裂解液重悬细菌,置冰上10min。
[5].高速离心后移出上清液,将上清液标记为B,即周质部分。
[6].在沉淀中加入100μl 0.1mM Tris-HCl (pH8.0)重悬沉淀,经干冰和37℃水浴反复冻溶3次,高速离心后移出上清液,将上清液标记为C,可溶性胞质部分。
[7].在沉淀中加入100μl 0.1M Tris-HCl (pH8.0)重悬沉淀,标记为D,即包涵体部分。
[8].向A中加入100μl 1×SDS上样缓冲液,B、C、D三部分各加入100μl 2×SDS上样缓冲液,100℃煮沸10min,4℃冷却。
[9].按上面相同方法配制12% SDS-PAGE进行样品电泳。
[10].按下面的方法对凝胶进行考玛斯亮蓝染色或银染分析。
3、蛋白质可视化:
[1].考马斯亮蓝染色
A.固定及染色:用至少5倍体积的考马斯亮蓝染色液浸泡凝胶,放摇床上室温缓慢旋转4h以上。回收染色液。
B.脱色:将凝胶浸泡在脱色液 [90ml甲醇:水(1:1) 溶液和10ml冰醋酸混合]中,缓慢摇动4~8小时脱色。其间换脱色液3~4次。直到脱色充分。
注意:脱色越充分,蛋白检测下限越低,脱色24h通常能检测到单一条带中含量低于0.1μg的蛋白质。
C.用凝胶成像系统在白屏上进行照相记录。也可将凝胶装入盛有20%甘油水溶液的塑料袋中,封闭保存。还可以将凝胶干燥成胶片保存。
D.考马斯亮蓝染色的胶,通过50% 甲醇、50 mM 碳酸氢铵(ammonium hydrogen carbonate)洗涤,再经过数次水洗后,可除去蛋白结合的考马斯亮蓝染料,能再用于银染色。
[2].银染色
A.固定:SDS-PAGE胶在至少5倍凝胶体积的乙醇:冰乙酸:水(30:10:60)的混合液固定浸泡4~12h;期间在摇床上平缓摇动。
B.倾去固定液,加入不少于5倍凝胶体积的30%乙醇,在室温下平缓摇动30min。
C.更换新鲜的30%乙醇,再平缓摇动30min。
D.倾去乙醇液,加入10倍凝胶体积的去离子水,在室温下平缓摇动10min。
E.更换去离子水,再平缓摇动10min。
F.染色:倾去去离子水,加入5倍体积的0.1% 硝酸银溶液(从20%的储存液新鲜稀释),在室温下平缓摇动30min。
G.倾去硝酸银溶液,用去离子水充分漂洗凝胶的两面,每次20s。期间防止凝胶表面干燥。
H.显影:加入5倍凝胶体积的新鲜配制的显影液(developer)(含2.5%碳酸钠和0.02% 甲醛的水溶液。配制:6.25g碳酸钠、100μl甲醛、250ml去离子水。),在室温下平缓摇动。仔细观察凝胶,数分钟内出现蛋白染色带,继续温育,直到所需的对比度。
I.终止:用1%乙酸洗涤凝胶数分钟,终止显色反应。
J.用去离子水洗涤凝胶数次,每次10min。胶在水中可保留数天。
K.用凝胶成像系统在白屏上进行照相记录。
注意事项:
[1].丙烯酰胺:丙烯酰胺具有很强的神经毒性,并可通过皮肤吸收,其作用具有累积性。称量时戴手套和面具。
[2].丙烯酰胺单体贮液在储存过程中因光和碱催化发生脱氨反应,会缓慢转化成丙烯酸和双丙烯酸,储存液应测定pH值不超过7.0。丙烯酰胺单体贮液应在暗色瓶中保存,每隔数月须重新配制。
[3].三氯乙酸:强腐蚀性。
[4].SDS(十二烷基硫酸钠):微细晶粒易于扩散,称量时戴面罩。
[5].蛋白质银染注意事项:
A.银染色要在塑料器皿中进行。
B.银染色中配制试剂和洗涤用的水一定是去离子水。
C.银染色中避免手、镊子等触及凝胶,以防造成污迹及斑点。
D.银染色中使用试剂应临用前新鲜配制。
E.固定时间较长,则加一步水洗30min,以免胶太脆而破碎。
F.甲醛在使用前加入。
G.第一次显色到溶液变混浊时应更换显色液,显色到条带清晰。
H.显色过程很快,要注意把握时间,避免染色过度。
I.银染液和显色液需要预冷。
[6].参考文献:
de Moreno MR, Smith JF, Smith RV. Mechanism studies of coomassie blue and silver staining of proteins. J Pharm Sci. 1986,75(9):907-11.
Congdon RW, Muth GW, Splittgerber AG.The binding interaction of Coomassie blue with proteins. Anal Biochem. 1993 Sep;213(2):407-13.
示例图片:

图12-9 SDS-PAGE analysis of Actinidia fruit juice proteins of various cultivars(From: Adv. Food Nutr. Res., 2007, 52: 293-324)
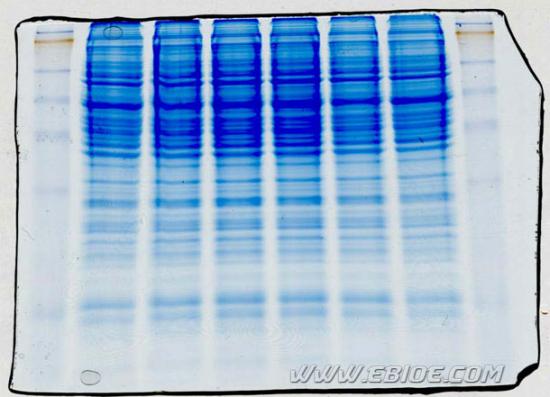
图12-10 SDS-PAGE protein gel stained with Coomassie blue
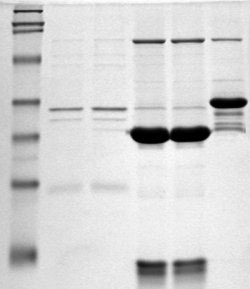
图12-11 SDS-PAGE protein gel stained with Coomassie blue

图12-12 SDS-PAGE protein gel stained with Coomassie blue

图12-13 silver-stained SDS-PAGE gels
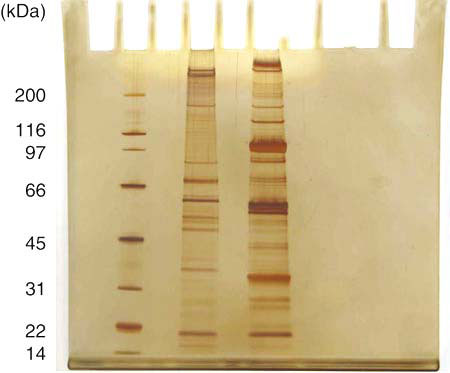
图12-13 silver-stained SDS-PAGE gels

图12-14 Proteins in two silver-stained SDS-PAGE gels
left: gel stored for two days, right: same gel after storage for 15 years.






