Applied and Environmental Microbiology, June 2004, p. 3618–3623
Axel Fey,1 Stefan Eichler,1 Se´bastien Flavier,2 Richard Christen,2 Manfred G.
摘要
定量PCR(Q-PCR)是一种对目的基因定量的快速高效的方法。在真核细胞里,定量-反转录-PCR(Q-RT-PCR)也在采用稳定表达的看家基因做参照的情况下用于基因表达的检测。在细菌体内,由于不存在稳定表达的看家基因,因此只有选择合适的RNA标准品才能对菌体内的RNA进行准确定量。本文建立了一种不仅能定量目的基因拷贝数(如细菌细胞数目),还可确定RNA拷贝数的精确定量方法,并用编码伤寒沙门氏菌invA和16S rRNA的基因来对该方法做出评价。目的基因的扩增片段用来做DNA的标准品,而同样的DNA标准品在体外转录用来做合适的RNA标准品,接种沙门氏菌的培养基和环境水样被用来做最终的检测。结果表明,运用Q-PCR和Q-RT-PCR 法均能对目的基因和RNA分子进行灵敏、精确、高重复性的定量。这是RNA标准被首次运用到原核生物的精确定量,表明这是一种对环境样品致病菌的存在性和代谢活性分析的新型检测方法。
前 言
水源中病源微生物的存在情况与人类健康息息相关(21,28)。传统的细菌培养检测方法不仅耗时,而且无法确定细菌的存活情况;PCR方法的应用便使上述问题得到了解决,并由此发展出多种针对水体病源微生物的检测方法(7,9,13)。但由于DNA在病原体死亡后还有很长的存在期,由此会造成假阳性的结果,因而在确立鉴定目的基因存在与否就代表细菌存在与否这个问题上就出现了争议(6)。因此,监测相对不稳定的RNA样本就成了另一个更为有效的方法(20)。
实时定量 PCR(Q-PCR)和反转录PCR(Q-RT-PCR)可以对基因、基因产物、甚至环境样品进行有效定量(16,24)。然而,由于缺乏可靠的参照,RNA精确定量的发展受到了局限。在真核细胞内,稳定表达的看家基因(如β-actin)可用作基因表达的相对定量的标准。遗憾的是,在细菌体内,还没有发现上述可稳定表达的基因。一部分人认为,可以选取16SrRNA的总量作为功能性基因表达的参照(12,14)。然而,rRNA的表达是和细菌的生理状态相关的(5,18)。当我们检测一系列环境样品的基因表达情况时,细菌的生理状态(如每个细胞16SrRNA的表达水平)是完全未知的。并且,这种方法无法对目的基因拷贝数进行准确定量。另一部分人则认为,可采用基因组DNA或包含基因组DNA片断的质粒作Q-RT-PCR的参照(17,22,23)。
然而,使用DNA作为标准品没有考虑到在反转录这一步中反转录效率的问题,而这一步会导致反应的效率与真实值相比会降低了84%-98.6%。因此,采用DNA作参照标准往往会低估实际的靶分子含量。真核细胞可采用细胞内转录的RNA作参照避免上述的情况,而细菌也需要找到一种类似的方法去解决这个问题。
我们研究的目的就是为了寻找一种能对环境水样中病菌基因和RNA样本进行准确定量的分析方法。以沙门氏菌为模式生物,将invA和 16SrRNA的编码基因作为目标基因,并采用在无菌的培养基或水样中接种进行验证。结果首次表明,体内转录的RNA标准品能成功地用于细菌内RNA的拷贝数的准确定量。这种以Q-PCR为基础的方法便能够对环境水样中病原体微生物的存在及代谢活动做出灵敏而准确的评估。
表1. DNA和RNA 标准品以及Q-PCR的对应引物

a T7启动子的相应序列用下划线标识,b 存在于基因的具体位置用数字范围表示
实验材料和方法
菌株和生长条件
沙门氏菌 ATCC14028置于心脑浸出液培养基中在37℃摇瓶培养(100rpm)。通过测量600nm的光密度值对菌体的生长情况进行监测。为了统计活细菌数目,样品经BHI梯度稀释后取50ul涂于BHI琼脂板上生长。37℃孵育24h后,统计每毫升的CFU值,仅选取克隆数为20-300所对应的稀释度进行计数。
核酸提取
按照厂商提供的方法 (QIAGEN, Hilden,Germany),采用DNeasy和Rneasy试剂盒分别对DNA和RNA进行提取。对于RNA提取,需加入 100ul的溶菌酶(500 ug/ ml-1)室温消化5min。在室温下用DNA酶处理(QIAGEN) 30min。
Q-PCR 的引物设计及标准品设计
所有的引物均购买于MWG Biotech (Ebersberg, Germany). Q-PCR和Q-RT-PCR的目标序列为编码伤寒沙门氏菌invA和16S rRNA的基因以及对应的RNA。为了给目标序列精确定量,我们设计了DNA和RNA标准品。对下面两种情况我们设计了另一套引物:(i1) 引物分别位于第一套引物扩增序列的上游和下游(如长扩增片断);(ii) 上游引物包含T7启动子序列(表1)。这些引物用于扩增S. enterica的基因组DNA,方法如下:初始变性94℃4 min;94℃1min,58℃(invA)/60℃(16S rRNA), 72℃1min,30个循环;最后一步延伸72℃6min。PCR混和液(每个样品50ul体系)包含:5ul 10×PCR缓冲液(QIAGEN),3 mM MgCl2, 200uM dNTP (Invitrogen, Karlsruhe, Germany), 400 nM (每份) 引物, 0.2ul Taq 聚合酶(QIAGEN), 1ng基因组DNA。PCR产物采用PCR 纯化试剂盒提取(QIAGEN) 并作为Q-PCR的DNA标准品。为了得到RNA标准品, 我们用Riboprobe System-T7 (Promega)试剂盒来对纯化PCR产物进行体外转录。随后用DNase I (15 min, 37°C)进行消化,并用RNeasy minikit (QIAGEN)提取所得的RNA,提取中还在柱上经历了一次DNaseI消化(室温15 min)。转录产物用1%的甲醛电泳分析。DNA和RNA标准品分别用PicoGreen (双链DNA定量试剂盒) 和RiboGreen (RNA定量试剂盒)获得(27)。标准品用无核酸酶的水稀释后置于-20 (DNA)或-70°C (RNA)保存。
拷贝数的计算
Q-PCR标准品拷贝数计算如下:平均来说,1bp的双链DNA分子量约为660Da,而一个单链的RNA核苷约为 340Da(PCR applications manual, 2nd ed., Roche Diagnostics GmbH, Mannheim,Germany, 1999)。计算方程式如下:拷贝数/纳克=(n×mw)/(NL×10-9),其中n是标准品碱基对或核苷长度,mw是指每个碱基对或核苷的分子量,NL则为阿伏加德罗常数(6.02×1023分子数/摩)。
Q- PCR和Q-RT-PCR
Q-PCR过程中,把5ul的稀释样品加到20ulPCR混和液里,PCR混和液由2×SYBR Green PCR Master Mix (Applied Biosystems)制备而来,并添加引物至终浓度400 nM。循环反应参数如下:95°C 10 min;95°C 20 s,60 (invA)或65°C (16S rRNA gene) 1 min,50个循环;接下来再进行95°C 20 s,60 (invA)或65°C (16S rRNA gene) 1 min。确定熔解曲线时,65°C到94°C每20 s上升1°C。对于Q-RT-PCR,在Q-PCR之前则采用SYBR Green RT-PCR试剂(Applied Biosystems)以及随机引物按试剂盒说明合成cDNA。RNA标准品也按照相似的方法合成。反转录过程中,一些RNA样本没有充足的反转录酶以消除DNA污染。相对两步法来说,一步Q-RT-PCR扩增法采用了SYBR Green RT-PCR试剂和序列特异性引物。虽然两者在扩增效率上没有差别,但两步法Q-RT-PCR常用于需进一步研究的实验中(如cDNA的长期保存,用同样的cDNA模板对不同靶基因进行定量)。上述Q-PCR和Q-RT-PCR都是在RotorGene 2000 (Corbett)上完成的。
数据分析
为产生标准曲线,系列稀释的DNA或RNA标准品(1,000 pg to 0.001 fg)在每轮Q-PCR反应中得到量化,并通过RotorGene software, version 4.6绘出标准曲线。对于每一个标准品,其浓度和荧光信号增长高于背景或达到域值时对应的循环数(Ct值)一一对应。标准曲线的斜率通过下列公式确定反应的效率:efficiency = 10-1/slope-1。由此可见,扩增效率等于1表明每一次循环中产物得到成倍扩增。通过标准曲线,RotorGene的配套软件可计算出待测样品的初始模板量。通过这些数值,我们便可以算出每毫升培养基或接种水样中模板拷贝数。
>
实验设计
为了绘制沙门氏菌的生长曲线,在盛有80 ml BHI培养基的250-ml摇瓶中接种2ml过夜培养产物(最终OD600=0.065)。每个管做3份平行,并置于37°C摇瓶培养(100 rpm),按不同的时间间隔测量样品的OD600以及活细胞计数(克隆计数)。核酸提取中,0.2到1ml的液体培养基(最多109个细菌细胞)被转入1.5ml反应管;RNA样品则被均匀分散到2倍体积的RNA保护剂(QIAGEN)中,室温放置5min以稳定细菌RNA (1),再经13,000×g离心10min,取上清冻存于-20°C备用。另外,在新鲜的BHI培养基中加入沙门氏菌的过夜培养产物,经4.5 h生长后(37°C, 100 rpm, 最终OD600=1.7),3,000×g离心10min后把沉淀悬浮于 0.25×Ringer 溶液(Oxoid)中,OD600被调整到0.2以使得每毫升培养液约含108个细胞。把1毫升上述培养物加入100 ml自来水或本研究院鱼池水中(每毫升培养液约含106个细胞),再把样品转入含90ml相应水样的瓶中,使菌密度为每毫升1-106个细胞。
结果分析
通过设计沙门氏菌invA和16S rRNA基因对应的特异引物,并采用Q-PCR和Q-RT-PCR方法对新的DNA和RNA参照进行了评价(Table 1)。每组样本都呈现出良好的线性标准曲线,相关系数(R2)可达到≥0.995,针对invA的线性范围达8个数量级,对16SrRNA则达7个数量级(Fig. 1)。在检出极限上,invA可达2个拷贝,16S rRNA则为1,000拷贝(Table 2)。反应效率值居于0.8和1.0之间(Table 2)。再经熔解曲线分析发现,每轮Q-PCR显示出很清晰的熔解曲线峰,而没有其他的非特异产物。
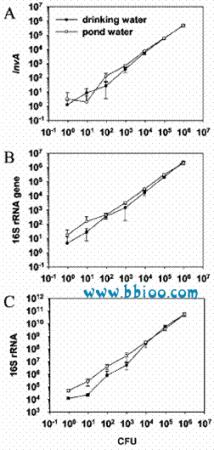
FIG.1 invA和16S rRNA对应的DNA(A)或RNA(B)标准品所绘出的标准曲线。图中各点体现了Ct值和目标核酸浓度的对应关系。Ct值是指荧光信号达到预设荧光域值时样本扩增所对应的循环数,域值是软件自动计算得出的。
采用纯培养物确证实验方法
首先对培养的菌体进行5天的监控,接种后30 min便进入了对数生长期,这个时期会一直持续2.5h。3天后,CFU数目开始减少(Fig. 2A)。不同时间点取样并制备核酸样品,再通过Q-PCR和Q-RT-PCR分别对invA和 16S rRNA的基因及其转录产物(RNAs)进行定量。在对数增长期,每CFU所对应16S rRNA和invA的基因拷贝数出现了增加(如接种后1.1 h),这是由染色体复制引起的(Fig. 2B)。接着,对应invA和16S rRNA每CFU拷贝数分别降低到1拷贝和5-10拷贝。这个结果印证了invA在沙门氏菌属中是单拷贝基因,它们有7个rrn操纵子(11)。试验中,16S rRNA和invA基因的比例大致恒定,一个invA对应8个16S rRNA拷贝 (Fig. 2C),这和预期值是一致的。RNA拷贝数则从接种后的2×104增加到对数期的最大值5×105(Fig. 2D)。随后,16S rRNA的拷贝数又会降低到刚接种状态。毒素基因invA在早对数期也有表达(最大值可达10 RNA拷贝 /CFU; Fig. 2D)。
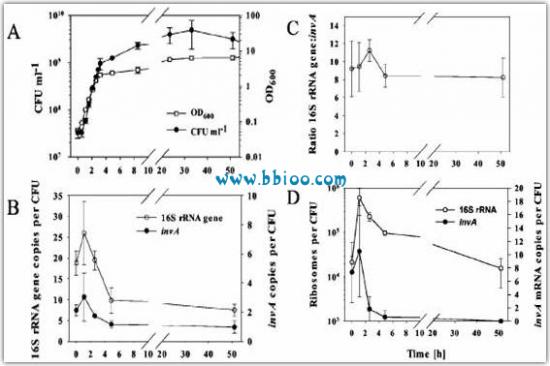
FIG.2. 沙门氏菌(ATCC14028)在37°C于BHI培养基中的生长曲线。(A) 不同时段采集样品的OD600s 和CFU. (B)根据Q-PCR所得每CFU对应的16S rRNA和invA的基因拷贝数. (C) 通过Q-PCR得出的 16S rRNA和invA的比率. (D)通过Q-RT-PCR得到每CFU对应的(16S rRNA)或invA mRNA. 所有数据都是3次平行反应所获取的平均值。标准偏差由图上的垂直短线标注。请注意x轴的间断处。
FIG. 3. 接种沙门氏菌的饮用水和池塘水样的Q-PCR和Q-RT-PCR分析。
(A)通过Q- PCR,每毫升CFU数目和invA拷贝数的对应关系。(B) 通过Q-PCR,每毫升CFU数目和16S rRNA基因拷贝数的对应关系。(C) 通过Q-PCR,每毫升CFU数目和16S rRNA拷贝数的对应关系。所有的接种水样测得数据都是3次平行反应获取的平均值。标准偏差由图上的垂直短线标注。
采用接种沙门氏菌的水样评估实验方法
对接种1-106/ml 沙门氏菌的饮用水和池塘水样进行分析,发现对于invA和16S rRNA每CFU具备相同的DNA拷贝数 (Fig. 3A and B)。细胞数目越少,越容易出现偏差,表明随着污染物水平的降低这一方法的精确度会降低。invA和 16S rRNA每CFU的基因拷贝数对于饮用水分别为0.69和2.9,对于池塘水则分别为0.74和 5.8。当水中接种104到106细胞时,饮用水和池塘水样对应的16S RNA表达很相似(Fig. 3C)。然而,较低的接种数目显示饮用水样中16S RNA表达低。综合来说,每CFU对应 16S rRNA的RNA拷贝数在饮用水和池塘水中分别为2.6×104和4.2×104。1拷贝inv基因对应16S rRNA 的平均拷贝数在饮用水和池塘水样中分别为5×104和5.5×104。对于未接种的对照水样,invA没有扩增信号,而16S rRNA (RNA 和 DNA)却显示出具1 CFU/ml水样同样背景值
实验结果与讨论
Q-PCR是环境水样细菌检出和定量检测的新型方法 (16,19)。定量方法建立在确定基因拷贝数的基础上,我们常采用基因组DNA来做标准品。但是,对于目的基因转录产物的定量这种方法便不可行了。在真核细胞中,我们可选取稳定表达的看家基因避免上述问题(3)。可是,细菌中并不存在这种稳定表达的看家基因。于是,我们发明了一种以Q-PCR为基础的定量方法,它不仅可以对细菌DNA精确定量,还可对环境水样中缺乏稳定表达看家基因的样本的RNA进行定量。据了解,这是首次成功的对原核细胞RNA进行定量。针对硅藻和海面植物的rbcL和rbcL基因的mRNA,我们也采用同样的方法用RNA标准品对其定量(25)。体外转录RNA被用做竞争性的RT-PCR的参照(29)。然而,采用随机引物容易造成RNA拷贝数的估测过高,原因是合成的短cDNA标准品缺乏引物结合位点。如果在Q-PCR过程中使用上述类似的引物合成DNA和RNA,也会出现相同的问题。虽然上述结果也可以用作Q-PCR的分析,但Q-RT-PCR过程中标准品的高估会造成对模板RNA拷贝数的估测高出实际值,这便使得此方法无法达到精确定量的要求。这是由于cDNA片断末端的不完整,和特异性引物结合位点缺失所造成的。为了避免上述问题,我们采用另一套引物用于RNA标准品的制备,这种引物扩增的片段比Q-PCR中得出的片段长(Table 1; Fig. 1)。其中,末端重叠是非常关键的,这使得合成cDNA的过程中的随机引物可得到延伸。另一个要考虑的问题是,DNase处理后DNA模板的残留 (10)。为了减小此因素的影响,我们通过再次DNase处理使得RNA标准品中残留DNA浓度减低4个数量级。
在纯净培养基和水样接种相关实验中(Fig. 2和3), 目标DNA可被精确定量。采用培养基接种,单个细胞的 16S rRNA和invA基因拷贝数均比预期值高4倍(Fig. 2B).这是由于在对数增长期染色体会出现加倍的情况。在此快速扩增期之后,一拷贝的invA约对应8拷贝的16S rRNA基因。这和预期值是相符的,表明invA是单拷贝基因并在沙门氏菌有7个rrn操纵子(11)。因此,沙门氏菌的DNA的回收率约为100%(如样品离心,细胞裂解,DNA提取和纯化)。对于接种样品,回收率约为70%(如0.7拷贝的invA 和2.9—5.8拷贝的16S rRNA基因)。但现今研究中,从相同水样中提取革兰氏阴性菌的效率从未超过52%。核糖体16S rRNA的拷贝数在对数早期增加很快(18), 而实验的后期却出现了下降。每CFU的16S rRNA拷贝数界于 20,000到500,000。核糖体数目和细菌的生长生理状况息息相关,单个细菌细胞的核糖体数目为6,700到71,000(2, 5, 8),和上述结果相符。既然纯化的小RNA被作为外参,那么16S rRNA的拷贝数就不会被过高评价。实际上,参照比样品的反转录效率以及后续的Q-PCR效率都要高(低估的可能性比高估的可能性要大)。因而,在复合培养基(如, BHI)中生长极快,得出的拷贝数稍高于实际值。采用基因组DNA做参照对葡萄球菌16S rRNA的研究表明,每拷贝16S rRNA基因对应35个16S rRNA的拷贝(23)。但是,Q- RT-PCR中一般不采用DNA作为参照,因为反转录的效率会在90%-1.4(4, 15)。这会造成目标RNA的过低估计。接种水样中的 16S rRNA表达率和培养于纯净培养基静止期表现相同(Fig. 3C)。但接种少量菌体的饮用水样的对应值比较低。原因是菌体在饮用水培养条件下无法达到适宜的生长状态。另一方面,核酸的提取和纯化也是一个限制因素。
本文以Q-PCR为基础,建立了一种不仅可以对DNA,还可以对细菌RNA进行精确定量的实验方法。细菌的生理生化状态是和每个细胞16S rRNA的数目息息相关的。这种新的方法被应用到环境样品的定量,样品活性监测,以及采用合适的靶序列,检测细菌的生理状态及潜在致病性。
参考文献
1. Bhagwat, A. A., R. P. Phadke, D. Wheeler, S. Kalantre, M. Gudipati, and M.
Bhagwat. 2003. Computational methods and evaluation of RNA stabilization reagents for genome-wide expression studies. J. Microbiol. Methods 55:399–409.
2. Bremer, H., and P. P. Dennis. 1996. Modulation of chemical composition and other parameters of the cell by growth rate, p. 1553–1569. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B.Magasanik, W. S. Reznikoff, M. Riley, M. Schaecter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, D.C.
3. Bustin, S. A. 2000. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 25:169–193.
4. Freeman, W. M., S. J. Walker, and K. E. Vrana. 1999. Quantitative RT-PCR:
pitfalls and potential. BioTechniques 26:112–125.
5. Gourse, R. L., T. Gaal, M. S. Bartlett, J. A. Appleman, and W. Ross. 1996. rRNA transcription and growth rate-dependent regulation of ribosome synthesis in Escherichia coli. Annu. Rev. Microbiol. 50:645–677.
6. Keer, J. T., and L. Birch. 2003. Molecular methods for the assessment of bacterial viability. J. Microbiol. Methods 53:175–183.
7. Kingombe, C., G. Huys, M. Tonolla, M. Albert, J. Swings, R. Peduzzi, and T.
Jemmi. 1999. PCR detection, characterization, and distribution of virulence genes in Aeromonas spp. Appl. Environ. Microbiol. 65:5293–5302.
8. Kjeldgaard, N. O., and K. Gausing. 1974. Regulation of biosynthesis of ribosomes, p. 369–392. In M. Nomura, A. Tissie`res, and P. Lengyel (ed.), Ribosomes. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
9. Malorny, B., J. Hoorfar, C. Bunge, and R. Helmuth. 2003. Multicenter validation of the analytical accuracy of Salmonella PCR: towards an international standard. Appl. Environ. Microbiol. 69:290–296.
10. Matthews, J., M. Chung, and J. Matyas. 2002. Persistent DNA contamination in competitive RT-PCR using cRNA internal standards: identity, quantity, and control. BioTechniques 32:1412–1417.
11. McClelland, M., K. E. Sanderson, J. Spieth, S. W. Clifton, P. Latreille, L. Courtney, S. Porwollik, J. Ali, M. Dante, F. Du, S. Hou, D. Layman, S. Leonard, C. Nguyen, K. Scott, A. Holmes, N. Grewal, E. Mulvaney, E. Ryan, H. Sun, L. Florea, W. Miller, T. Stoneking, M. Nhan, R. Waterston, and R. K. Wilson. 2001. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413:852–856.
12. Neretin, L. N., A. Schippers, A. Pernthaler, K. Hamann, R. Amann, and B. B. Jorgensen. 2003. Quantification of dissimilatory (bi)sulphite reductase gene expression in Desulfobacterium autotrophicum using real-time RT-PCR. Environ. Microbiol. 5:660–671.
13. Paton, A., and J. Paton. 1998. Detection and characterization of Shiga toxigenic Escherichia coli by using multiplex PCR assays for stx1, stx2, eaeA, enterohemorrhagic E. coli hlyA, rfbO111, and rfbO157. J. Clin. Microbiol. 36:598–602.
14. Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [Online.]
15. Pfaffl, M. W., and M. Hageleit. 2001. Validities of mRNA quantification using recombinant RNA and recombinant DNA external calibration curves in real-time RT-PCR. Biotechnol. Lett. 23:275–282.
16. Rodriguez-Lazaro, D., M. Hernandez, T. Esteve, J. Hoorfar, and M. Pla. 2003. A rapid and direct real time PCR-based method for identification of Salmonella spp. J. Microbiol. Methods 54:381–390.
17. Rokbi, B., D. Seguin, B. Guy, V. Mazarin, E. Vidor, F. Mion, M. Cadoz, and M.-J. Quentin-Millet. 2001. Assessment of Helicobacter pylori gene expression within mouse and human gastric mucosae by real-time reverse transcriptase PCR. Infect. Immun. 69:4759–4766.
18. Ruimy, R., V. Breittmayer, V. Boivin, and R. Christen. 1994. Assessment of the state of activity of individual bacterial cells by hybridization with a ribosomal RNA targeted fluorescently labelled oligonucleotidic probe. FEMS Microbiol. Ecol.. 15:207–213.
19. Sharma, V. K., E. A. Dean-Nystrom, and T. A. Casey. 1999. Semi-automated fluorogenic PCR assays (TacMan) for rapid detection of Escherichia coli O157:H7 and other Shiga toxigenic E. coli. Mol. Cell. Probes 13:291–302.
20. Sheridan, G. E. C., C. I. Masters, J. A. Shallcross, and B. M. Mackey. 1998. Detection of mRNA by reverse transcription-PCR as an indicator of viability in Escherichia coli cells. Appl. Environ. Microbiol. 64:1313–1318.
21. Straub, T. M., and D. P. Chandler. 2003. Towards a unified system for detecting waterborne pathogens. J. Microbiol. Methods 53:185–197.
22. Vandecasteele, S. J., W. E. Peetermans, R. Merckx, and J. Van Eldere. 2001. Quantification of expression of Staphylococcus epidermidis housekeeping genes with Taqman quantitative PCR during in vitro growth and under different conditions. J. Bacteriol. 183:7094–7101.
23. Vandecasteele, S. J., W. E. Peetermans, R. Merckx, M. Van Ranst, and J. Van Eldere. 2002. Use of gDNA as internal standard for gene expression in staphylococci in vitro and in vivo. Biochem. Biophys. Res. Commun. 291: 528–534.
24. Walker, N. J. 2002. A technique whose time has come. Science 296:557–559.
25. Wawrik, B., J. H. Paul, and F. R. Tabita. 2002. Real-time PCR quantification of rbcL (ribulose-1,5-bisphosphate carboxylase/oxygenase) mRNA in diatoms and pelagophytes. Appl. Environ. Microbiol. 68:3771–3779.
26. Weinbauer, M. G., I. Fritz, D. F. Wenderoth, and M. G. Ho¨fle. 2002. Simultaneous extraction from bacterioplankton of total RNA and DNA suitable for quantitative structure and function analyses. Appl. Environ. Microbiol. 68:1082–1087.
27. Weinbauer, M. G., and M. G. Ho¨fle. 2001. Quantification of nucleic acids from aquatic environments by using green-fluorescent dyes and microtiter plates, p. 2.1.3–2.1.10. In A. Akkermans, J. van Elsas, and F. de Bruijn (ed.), Molecular microbial ecology manual, 5th suppl. Kluwer Academic Publishers, Dordrecht, The Netherlands.
28. World Health Organization. 2002. The world health report. Reducing risks, promoting healthy life. World Health Organization, Geneva, Switzerland.
29. Zhang, J., and C. D. Byrne. 1999. Differential priming of RNA templates during cDNA synthesis markedly affects both accuracy and reproducibility of quantitative competitive reverse-transcriptase PCR. Biochem. J. 337:231– 241.






