本文转自医药魔方数据微信,发布已获医药魔方授权,如需转载,请与医药魔方联系。
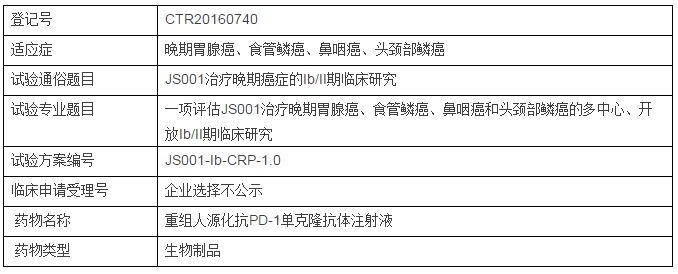
根据药物临床试验登记与信息公司平台9月28日首次显示的信息,君实生物即将启动PD-1单抗JS001治疗晚期胃腺癌、食管鳞癌、鼻咽癌、头颈部鳞癌的I/II期临床研究。
该项单臂、开放标签、非随机研究于2016年9月14日获得中山大学肿瘤防治中心伦理委员会审查通过,计划入组326例患者。
上述信息提示,君实生物的PD-1单抗将领先其他国内公司同类产品,率先进入II期临床阶段。该研究目前的状态是“进行中,尚未招募”。
临床试验信息
试验目的
初步评价重组人源化抗PD-1单克隆抗体注射液(JS001)治疗晚期胃腺癌、食管鳞癌、鼻咽癌和头颈部鳞癌的抗肿瘤活性,并确定II期临床研究推荐剂量(RP2D)
受试者信息
年龄:18岁(最小年龄)至75岁(最大年龄)
性别:男+女
健康受试者: 无
入选标准
1. 对本研究已充分了解并自愿签署知情同意书(ICF);
2. 经组织学和/或细胞学确诊的晚期和/或转移性胃腺癌(含胃食管结合部腺癌)、食管鳞癌、鼻咽癌、头颈部鳞癌患者;
3. 对于胃腺癌患者,既往接受过至少一种针对晚期胃腺癌的药物治疗且确定肿瘤进展或不耐受现有的化疗方案;如果根治性手术伴随的辅助或新辅助化疗结束后6个月内复发或转移,可以入组本项研究;
4. 对于食管鳞癌患者,既往接受过至少一种针对晚期食管鳞癌的治疗(包括但不限于抗肿瘤药物治疗、放疗或联合放化疗)且确定肿瘤进展或不耐受现有的化疗方案;如果根治性手术伴随的辅助或新辅助治疗(包括但不限于化疗、放疗、联合放化疗)结束6个月内复发或转移,可以入组本项研究;
5. 对于鼻咽癌、头颈部鳞癌的患者,既往接受过至少一种针对晚期鼻咽癌或头颈部鳞癌的治疗(包括但不限于抗肿瘤药物治疗、放疗或联合放化疗)且确定肿瘤进展或不耐受现有的其它治疗方案;如果根治性手术伴随的辅助或新辅助同步放化疗结束后6个月内复发或转移,可以入组本项研究。
6. 至少有一个可测量病灶(根据RECIST1.1); 注:之前接受过放疗的病灶不可以视为靶病灶,除非放疗后病灶发生明确进展。
7. 同意提供既往储存的肿瘤组织标本或者进行活检以采集肿瘤病灶组织送往中心实验室进行PD-L1 IHC检测。胃腺癌、食管鳞癌和头颈部鳞癌的患者,PD-L1表达必须为阳性(阳性定义为采用Spring Bioscience PD-1/L1 IHC测试试剂,PD-L1表达>1%)方可以入组;
8. 年龄18岁~75岁,性别不限;
9. ECOG评分0-1分;
10. 预期生存期≥3月;
11. 入组前7天内实验室检查值必须符合相关标准:
12. 入组前21 天内,育龄期女性必须确认血清妊娠试验为阴性并同意在研究药物使用期间以及最后一次给药后28 天内采用有效避孕措施。本方案中育龄期女性定义为性成熟女性:1)未经历子宫切除术或双侧卵巢切除术,2)自然停经未持续连续的24 个月(癌症治疗后闭经不排除有生育能力)(即,在之前连续的24个月内的任何时间出现过月经)。
排除标准
1. 已知对一水枸橼酸、二水枸橼酸钠、甘露醇、聚山梨酯过敏者(试验药物的组分);
2. 入组前4周内接受过抗肿瘤的细胞毒化学药物治疗、生物药物治疗(如单克隆抗体)、免疫治疗(如白介素2或干扰素),或其它研究药物治疗,或仍处于此类药物的5个半衰期以内;
3. 入组前2周内接受过酪氨酸激酶抑制剂治疗;
4. 入组前4周内接受过放疗,或8周内接受过放射性药物治疗,但针对骨转移病灶的局部姑息性放疗除外;
5. 入组前4周内进行过外科大手术或尚未从之前的手术中完全恢复(外科大手术的定义参照2009年5月1日施行的《医疗技术临床应用管理办法》中规定的3级和4级手术);
6. 既往抗肿瘤治疗的毒性反应尚未恢复到CTCAE[4.03版]0-1 级,以下情况除外:a.脱发;b. 色素沉着;c.周围神经毒性已恢复到≤CTCAE2级;d.放疗引起的远期毒性,经研究者判断不能恢复;
7. 具有临床症状的中枢神经系统转移(如脑水肿、需要激素干预,或脑转移进展)和/或癌性脑膜炎的患者。既往接受过脑或脑膜转移治疗,如临床稳定已维持至少2 个月,并且已经停止全身性激素治疗(剂量>10mg/天的泼尼松或其它等疗效激素)大于4 周的患者可以纳入;
8. 鼻咽癌和头颈部鳞癌患者入组前4周内检查发现有坏死性病灶,研究者判断有大出血风险;
9. 入组前4周内接受过全身或局部的糖皮质激素治疗;
10. 既往或现在同时患有其它恶性肿瘤(除了得到有效控制的非黑色素瘤的皮肤基底细胞癌、乳腺/宫颈原位癌、和其它在过去五年内没有治疗也得到有效控制的恶性肿瘤);
11. 患者存在任何活动性自身免疫性疾病或有自身免疫性疾病病史(包括但不限于:间质性肺炎、葡萄膜炎、肠炎、肝炎、垂体炎、肾炎、甲状腺功能亢进、甲状腺功能降低;患有白癜风或在童年期哮喘已完全缓解,成人后无需任何干预的患者可纳入;患有需要支气管扩张剂进行医学干预的哮喘则不能纳入);
12. 既往使用过抗PD-1抗体、抗PD-L1抗体、抗PD-L2抗体或抗CTLA-4抗体(或作用于T细胞协同刺激或检查点通路的任何其它抗体);
13. 患有活动性肺结核(TB)的患者,正在接受抗结核治疗或者筛选前1年内接受过抗结核治疗;
14. 患有需要长期使用免疫抑制药物治疗、或需要全身或局部使用具有免疫抑制作用剂量的皮质类固醇的合并症;
15. 入组前4周内接种过任何抗感染疫苗(如流感疫苗、水痘疫苗等);
16. 妊娠或哺乳期妇女;
17. HIV阳性;
18. HBsAg阳性同时检测到HBVDNA拷贝数阳性(定量检测≥1000cps/ml);
19. 慢性丙型肝炎血液筛查阳性(HCV抗体阳性);
20. 研究者认为可影响方案依从性,或影响患者签署知情同意书(ICF),或不适宜参加本临床试验的具有临床意义的任何其它疾病或状况。
试验分组
试验药:
重组人源化抗PD-1单克隆抗体注射液3mg/kg剂量组,无菌水针剂型;240mg/6ml/瓶;使用在线过滤器(0.2或0.22μm)进行静脉滴注,在60分钟内进行静脉点滴;每2周给药一次;每4周一个治疗周期,直到无法继续获益、不可耐受的毒性、研究者决定、撤回知情同意或死亡;
重组人源化抗PD-1单克隆抗体注射液10mg/kg剂量组,无菌水针剂型;240mg/6ml/瓶;使用在线过滤器(0.2或0.22μm)进行静脉滴注,在60分钟内进行静脉点滴;每2周给药一次;每4周一个治疗周期,直到无法继续获益、不可耐受的毒性、研究者决定、撤回知情同意或死亡;
对照药:不适用
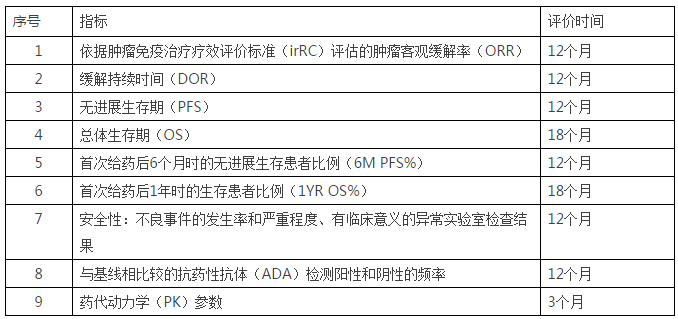
终点指标
主要终点:依据实体瘤疗效评价标准(RECIST 1.1)评估的肿瘤客观缓解率(ORR)。评价时间为12个月。
次要终点
研究者信息
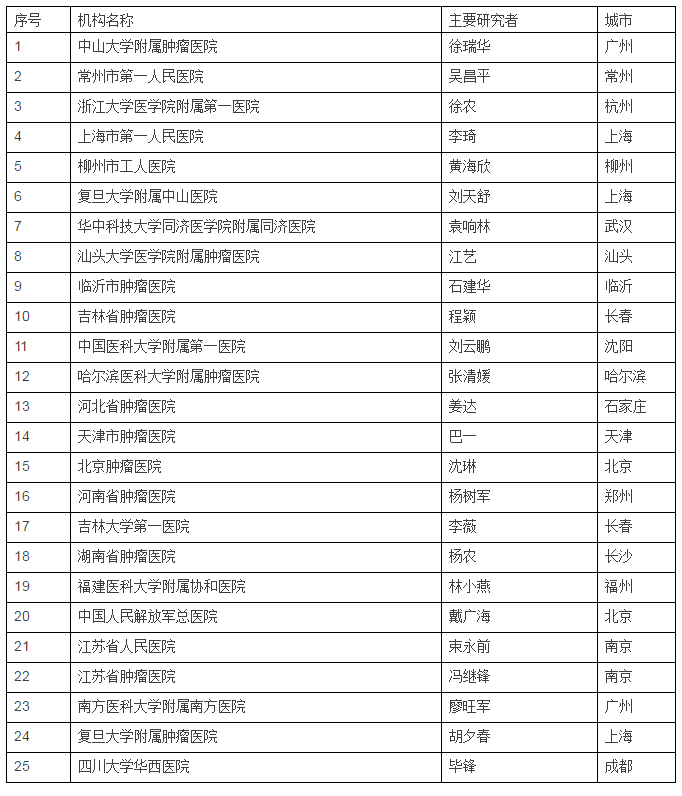
主要研究者
中国地区各参加机构