加州大学、圣地亚哥医学院和加州大学圣迭戈穆尔斯癌症中心的研究人员发现了新药研发方法,能够破坏高度分裂的肿瘤细胞。这项研究提出了一个高效、备用的方法,用于杀死快速生长的癌细胞,而不会引起目前疗法的一些副作用。相关研究发表在11月13日在线版的《Nature Medicine》期刊上。
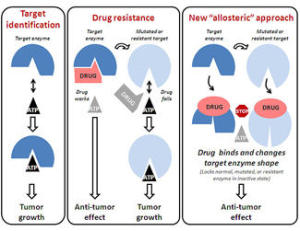
图:变构方法
任职于穆尔斯癌症中心病理学教授、转录研究所副主任的David A. Cheresh 博士领导这项研究,该小组的科学家使用一种创新性生化方法去研制一类新药物,通过结合并改变RAF酶的结构,能够抑制几乎所用肿瘤细胞的分裂。
RAF的研究已经开展很长一段时间,但是它在细胞分裂上的作用令人惊讶。Cheresh 博士称:“通过设计一类能够改变RAF结构的新药,我们揭示RNF在高分裂肿瘤细胞中的之前未被发现的新功能。”
目前,针对靶酶(如RAF)的肿瘤药物通常作用于酶的活性部位,然而,它们往往缺少特异性。Cheresh 博士称:“这类药攻击许多不同的靶点,意味着会引起预料之外的副作用和剂量受限的毒理特性。” 更多的关注是:肿瘤细胞往往形成对这类药物的抗性,使得它们不能杀死肿瘤细胞。
Cheresh 和同事开发出一类RAF抑制剂的新药,它们不能结合在酶的活跃位点,从而避免现有药品的限制。这类新药被称为变构抑制剂,改变靶酶的形状,进而让靶酶失效。经测试,特定新药—KG5能筛选出增殖细胞的RAF,却忽视正常或休眠的细胞。在药物作用的肿瘤细胞中,RAF不能结合在丝分裂器上用于介导细胞分裂,导致细胞分裂受限,最终引起细胞凋亡或程序性死亡。KG5以相同方式有效地干扰血管的增殖(这一个过程被称为血管生成)。
经测试,KG5在肿瘤细胞系、动物模型和人类肿瘤患者的组织切片中产生类似的结果。研究小组已经开发出KG5的改进型,其功效是原有药品的100倍。他们希望这些功效更强的化合物的其中一种将来能在穆尔斯癌症中心应用于临床试验。(生物探索译)
相关英文论文摘要:
A MEK-independent role for CRAF in mitosis and tumor progression
RAF kinases regulate cell proliferation and survival and can be dysregulated in tumors1, 2. The role of RAF in cell proliferation has been linked to its ability to activate mitogen-activated protein kinase kinase 1 (MEK) and mitogen-activated protein kinase 1 (ERK). Here we identify a MEK-independent role for RAF in tumor growth. Specifically, in mitotic cells, CRAF becomes phosphorylated on Ser338 and localizes to the mitotic spindle of proliferating tumor cells in vitro as well as in murine tumor models and in biopsies from individuals with cancer. Treatment of tumors with allosteric inhibitors, but not ATP-competitive RAF inhibitors, prevents CRAF phosphorylation on Ser338 and localization to the mitotic spindle and causes cell-cycle arrest at prometaphase. Furthermore, we identify phospho-Ser338 CRAF as a potential biomarker for tumor progression and a surrogate marker for allosteric RAF blockade. Mechanistically, CRAF, but not BRAF, associates with Aurora kinase A (Aurora-A) and Polo-like kinase 1 (Plk1) at the centrosomes and spindle poles during G2/M. Indeed, allosteric or genetic inhibition of phospho-Ser338 CRAF impairs Plk1 activation and accumulation at the kinetochores, causing prometaphase arrest, whereas a phospho-mimetic Ser338D CRAF mutant potentiates Plk1 activation, mitosis and tumor progression in mice. These findings show a previously undefined role for RAF in tumor progression beyond the RAF-MEK-ERK paradigm, opening new avenues for targeting RAF in cancer.
英文论文链接:https://www.nature.com/nm/journal/vaop/ncurrent/full/nm.2464.html






