猕猴是人类的近亲,也是在研究中最广泛使用的非人灵长类模式动物。虽然猕猴在生物学和医学方面的应用已经有很长的时间,但人们对猕猴基因组序列多态性的了解却非常有限。
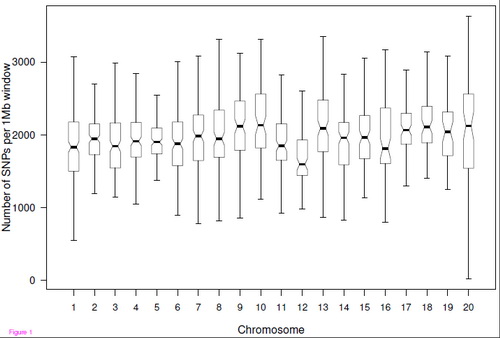
中国科学院昆明动物研究所宿兵实验室(博士研究生张雁峰等)和华大基因合作,采用二代测序的方法对中国猕猴进行了全基因组序列分析。测序深度为猕猴基因组11.56倍的覆盖率。通过和国外已测的印度猕猴基因组序列的比较,他们共发现了550万个单核苷酸序列多态性位点(SNP),其中294万个SNP是中国猕猴基因组中的杂合多态性,256万个SNP是中国猕猴和印度猕猴间的差异位点。同时,他们还发现了12万多个的基因组结构变异(Structural variation, SV)。
这项工作是首次对猕猴全基因组序列多态性的解析,所报道的海量序列多态数据将为以猕猴为对象的生物学和临床前医学研究提供具有重要价值的数据库。相关研究结果发表于基因组学的知名刊物Genome Biology(https://genomebiology.com/2011/12/7/R63/abstract)。
生物探索推荐英文原文:
Genome sequence and global sequence variation map with 5.5 million SNPs in Chinese rhesus macaque
Background
Rhesus macaque (Macaca mulatta) is the most widely used nonhuman primate animal in biomedical research. A global map of genetic variations in rhesus macaque is valuable for both evolutionary and functional studies.
Results
Using next-generation sequencing technology, we sequenced a Chinese rhesus macaque genome with 11.56-fold coverage. In total, 96% of the reference Indian macaque genome was covered by at least one read, and we identified 2.56 million homozygous and 2.94 million heterozygous SNPs. We also detected a total of 125,150 structural variations, of which 123,610 were deletions with a median length of 184 bp (ranging from 25bp to 10kb); 63% of these deletions were located in intergenic regions and 35% in intronic regions. We further annotated 5,187 and 962 nonsynonymous SNPs to the macaque orthologs of human disease and drug-target genes, respectively. Finally, we set up a genome-wide genetic variation database with the use of Gbrowse.
Conclusions
Genome sequencing and construction of a global sequence variation map in Chinese rhesus macaque with the concomitant database provide applicable resources for evolutionary and biomedical research.
The complete article is available as a provisional PDF. The fully formatted PDF and HTML versions are in production.






