摘要:国际学术期刊《公共科学图书馆—病原体》(PLoS Pathogens)在线发表了中科院上海生命科学研究院生化与细胞所王琛课题组的研究论文“Mitochondrial ubiquitin ligase MARCH5 promotes TLR7 signaling by attenuating TANK action”。该论文报道了线粒体泛素连接酶MARCH5能够特异性调控TLR7信号通路,首次将线粒体与TLR信号通路联系起来,揭示了线粒体在固有免疫促炎症反应调节中的关键作用。
在固有免疫信号通路中,TLR (Toll-like Receptor)、RIG-I等模式识别受体(Pattern Recognition Receptor; PRR)能够识别不同的病原体相关分子模式(包括真菌、细菌的特异性细胞壁组分,病毒的RNA等),PRR活化后通过一系列接头分子将信号向下游传递,活化IRF(3/7)、NF-kB等转录因子并启动转录,最终诱导一系列功能分子(包括细胞因子、趋化因子、ISGs等)的表达,启动固有免疫反应,实现细胞的免疫防御过程。已有的研究表明:RIG-I信号通路介导的抗病毒反应中,线粒体发挥至关重要的作用,一些信号通路分子之间的相互作用需要特定的线粒体组分参与才能够实现;TLR和RIG-I共用一部分下游信号通路,但并不清楚线粒体在TLR下游信号通路调节中是否具有功能。
通过对线粒体蛋白进行大规模筛选,课题组石贺欣、刘星等发现,泛素连接酶MARCH5可能调控固有免疫反应TLR7信号通路。实验证明,线粒体通过表达MARCH5显著地促进了TLR7介导的NF- kB调控的基因表达,而RNA干扰敲低内源MARCH5的表达则抑制NF- kB调控的基因表达;MARCH5能够特异性地与TANK蛋白分子结合。实验室过去的工作证明:TRAF6在固有免疫信号通路中扮演关键角色,它可以发生自身K-63位多聚泛素化,并促进NEMO(另一个关键节点分子)的K-63位多聚泛素化,对于TLR7介导的NF- kB通路激活十分关键。Akira实验室的工作证明:TANK能够和TRAF6相互作用,并抑制TRAF6的信号传递功能。本研究发现MARCH5催化了TANK的多聚泛素化(K63分枝形式),因而解除了TANK对TRAF6的抑制。有意思的是,MARCH5调控TLR7信号通路的功能依赖于它的线粒体定位,将MARCH5的线粒体定位序列突变,使其定位于细胞质中或者细胞膜上,则丧失了催化TANK泛素化的功能。
近三年来,王琛课题组在固有免疫信号通路调控领域开展了较系统性的研究,取得了阶段性的研究进展。发现了多个参与调控的新分子(UXT、Trim21、Herc5、Tom70、IFIT3、Cited2等),分别调控信号通路的不同层面;揭示了磷酸化、乙酰化、泛素化、ISG15化等多种蛋白质翻译后修饰在固有免疫反应信号通路中的调控作用;认识了多种蛋白分子之间特异性相互作用(Specific Protein Interaction)、蛋白质复合物动态形成(Signaling-dependent Complex Formation)以及蛋白分子的亚细胞空间定位(Subcellular Localization)等细胞生理过程如何实现信号的精细调控。部分工作结果已经发表在J Immunol、Mol Biol Cell、Mol Cell Biol、Cell Res、PLoS Pathog等杂志上。
上述系列研究工作得到了国家科技部、基金委 、中国科学院以及上海市科委等机构的经费支持。
生物探索推荐英文原文摘要:
PLoS Pathog 7(5): e1002057.
doi:10.1371/journal.ppat.1002057
Mitochondrial Ubiquitin Ligase MARCH5 Promotes TLR7 Signaling by Attenuating TANK Action
Abstract: The signaling of Toll-like receptors (TLRs) is the host's first line of defense against microbial invasion. The mitochondrion is emerging as a critical platform for antiviral signal transduction. The regulatory role of mitochondria for TLR signaling remains to be explored. Here, we show that the mitochondrial outer-membrane protein MARCH5 positively regulates TLR7 signaling. Ectopic expression or knockdown of MARCH5 enhances or impairs NF-κB-mediated gene expression, respectively. MARCH5 interacts specifically with TANK, and this interaction is enhanced by R837 stimulation. MARCH5 catalyzes the K63-linked poly-ubiquitination of TANK on its Lysines 229, 233, 280, 302 and 306, thus impairing the ability of TANK to inhibit TRAF6. Mislocalization of MARCH5 abolishes its action on TANK, revealing the critical role of mitochondria in modulating innate immunity. Arguably, this represents the first study linking mitochondria to TLR signaling.
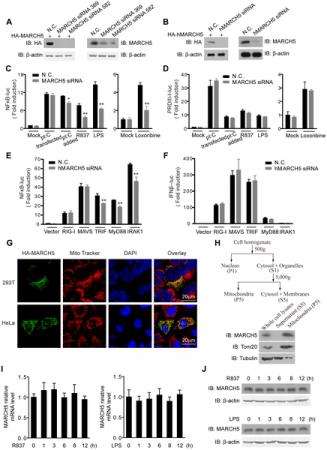
Pictured: A, HEK293T cells were transfected with HA-MARCH5 and then treated with the nonspecific control (N.C.) or MARCH5 siRNA (left panel). Raw264.7 cells were transfected with the negative control (N.C.) or MARCH5 siRNA (right panel). Cell lysates were immunoblotted with the indicated antibodies. B, HEK293T cells were transfected with HA-hMARCH5 and then treated with the nonspecific control (N.C.) or hMARCH5 siRNA (left panel). HEK293T cells were transfected with the negative control (N.C.) or hMARCH5 siRNA (right panel). Cell lysates were immunoblotted with the indicated antibodies. C and D, the nonspecific control (N.C.) or MARCH5 siRNA were transfected into Raw264.7 cells with NF-κB (C) or PRDIII-I reporter plasmids (D), respectively. Forty-eight hours after transfection, cells were stimulated with poly(I:C) (2 µg/ml, transfected), poly(I:C) (50 µg/ml, added to the culture medium), R837 (10 µg/ml), Loxoribine (50 µM) or LPS (100 ng/ml) for 6 h before luciferase assays were performed. A pTK-Renilla reporter was used to normalize data. E and F, the nonspecific control (N.C.) or hMARCH5 siRNA were transfected into HEK293T cells together with NF-κB (E) or IFNβ reporter plasmids (F), respectively. Forty-eight hours after transfection, cells were transfected again with RIG-I, MAVS, TRIF, MyD88 or IRAK1 for 16 h before luciferase assays were performed. A pTK-Renilla reporter was used to normalize data. G, HA-MARCH5 was transfected into HEK293T cells (upper panel) or HeLa cells (lower panel), which were then stained with an anti-HA antibody and imaged by confocal microscopy. The mitochondria were stained with MitoTracker. H, HEK293T cells were fractionated as shown in diagram (top panel). The corresponding fractions were analyzed by immunoblotting with the indicated antibodies (bottom panel). I and J, Raw264.7 cells were stimulated with R837 (10 µg/ml) or LPS (100 ng/ml), respectively, for the indicated time periods. The mRNA level of MARCH5 was measured by quantitative PCR (I), and the protein level of MARCH5 was measured by immunoblotting with an anti-MARCH5 antibody (J). Data from C–F and I are presented as means ± S.D. from three independent experiments. *, P<0.05; **, P<0.01.
In 2005, MAVS was characterized as the critical adaptor protein for the signal transduction of RIG-I-like receptors (RLRs). This provided the first link between mitochondria and the intracellular antiviral defense system. From then on, exploring the potential functions of novel mitochondrial proteins in microbe-host interactions became a rapidly expanding frontier. Notably, it remains unknown whether mitochondrial proteins can directly regulate TLR signaling. Here, we demonstrate that the mitochondrial protein MARCH5 positively modulates TLR7 signaling. Our study reveals that MARCH5 is a novel E3 ubiquitin ligase and catalyzes the K63-linked poly-ubiquitination of TANK. This modification releases the inhibitory effects of TANK on TRAF6. Arguably, this represents the first study linking mitochondria to TLR signaling, shedding new light on the role of mitochondria in the proinflammatory response.
A new paradigm has been established in the past decade, revealing how Toll-like receptors (TLRs) detect a wide range of pathogens and then initiate immediate host defenses. As a result, cytokines and chemokines are induced to mobilize immune cells for controlling and eliminating pathological infections. Given that the TLR signal transduction cascade is the first line of the host defense against pathogens, they are subjected to multiple layers of positive and negative regulations. Herein, we characterize the mitochondrial protein MARCH5 as an essential and positive modulator of TLR7 signaling.
In this study, several lines of evidence substantiate the novel function of MARCH5 in TLR7 signaling. First, exogenous expression of MARCH5 potentiated the induction of NF-κB responsive genes upon R837 stimulation, but not the induction of IRF3/7 responsive genes. Second, knockdown of MARCH5 unequivocally resulted in the reduction of NF-κB-mediated gene expression, and this attenuation was rescued by exogenously expressing a siRNA-resistant rMARCH5. Third, MARCH5 interacted with TANK, a negative regulator of TLR7 signaling. Interestingly, TANK could partially co-localize to mitochondria in response to TLR7 stimulation. This interaction was increased upon R837 challenge, suggesting that the interaction was transient and dynamic. Fourth, knockdown of TANK impaired the ability of MARCH5 to potentiate TLR7 signaling.
Previous in vitro studies suggested that TANK positively regulates TBK1 and IKKε-mediated production of type I interferon [36]. However, analysis of TANK−/− mice indicated that TANK is not essential for the induction of type I interferon downstream of RIG-I/MDA5 or TRIF [19]. Further analysis revealed that TANK is critical for the negative regulation of canonical NF-κB activation via suppression of TRAF6 auto-ubiquitination [19]. The underlying mechanism is still not clear.
Ubiquitination is an effective mechanism to regulate TLR signaling pathways. E3 ubiquitin ligases (Nrdp1, A20) and de-ubiquitinases (DUBA, CYLD, and A20) have been demonstrated as positive or negative modulators of these pathways. Nrdp1 ‘preferentially’ promotes TLR-mediated production of type I interferon [37]. A20 and CYLD ‘preferentially’ terminate TLR-induced activation of NF-κB by de-ubiquitinating their substrates, such as RIP1, TRAF6, TAK1, NEMO and so on [15], [16]. DUBA interacts with and de-ubiquitinates TRAF3, thereby attenuating TLR-dependent and TLR-independent antiviral responses [38].
K48-linked poly-ubiquitin chains usually target substrates for proteasome degradation, whereas K63-linked poly-ubiquitin chains usually regulate substrate activity but do not promote effective degradation. Gatot et al. previously reported that TANK is subjected to lipopolysaccharide mediated K63-linked poly-ubiquitination [35]. However, the identity of the relevant E3 ubiquitin ligase and the functional consequence of TANK poly-ubiquitination remained to be revealed. In this study, we showed that ectopic expression of MARCH5 enhanced poly-ubiquitination of TANK after R837 stimulation, whereas knockdown of MARCH5 attenuated this poly-ubiquitination. Wild type MARCH5 catalyzed the K63-linked poly-ubiquitination of TANK on its Lysines 229, 233, 280, 302 and 306. Neither MARCH5 C2A nor MARCH5 ΔRING could synergize the activation of NF-κB, stimulated by R837. The siRNA-resistant mutants (rMARCH5 C2A or rMARCH5 ΔRING) could not rescue the NF-κB activation in MARCH5 knockdown cells. In addition, TANK per se could interfere with TRAF6 auto-ubiquitination.
The in vitro ubiquitination assay revealed that MARCH5 could catalyze the formation of both K48- and K63-linked polyUb chains. This is somewhat unexpected given that MARCH5 mediates only K63-linked polyubiquitination of TANK in transfected cells. One possible explanation for this discrepancy is that additional cofactors may exist in vivo to guide the reaction of MARCH5 in favor of the K63 linkage. Indeed, we have previously shown that TRAF6 only catalyzes the formation of K63-linked polyUb chains in vivo, which positively regulate the NF-κB signaling pathway [39]. Similar to what is observed for MARCH5, TRAF6 could facilitate the assembly of both K48- and K63-linked polyUb chains in vitro [40]. Recent mechanistic studies of polyUb chain formation revealed that additional proteins were involved in the determination of polyUb linkage [41], [42]. Strikingly, the NEMO protein is reported to be modified in vivo by linear-, K27- and K63-linked polyUb chains under different physiological conditions [43]. It is thus a great challenge to monitor the dynamic formation of various polyUb chains of different linkages and determine their cognate functional consequences. Currently, we are trying to use the RNAi approach to screen for potential cofactor(s) of MARCH5. Hopefully, this will shed new light on how MARCH5 catalyzes the formation of K63-linked polyUb chains in vivo. Taken together, we propose that MARCH5 is an authentic E3 ubiquitin ligase and catalyzes K63-linked poly-ubiquitination of TANK. MARCH5 modulates TLR7 signaling via releasing the inhibitory action of TANK toward TRAF6.
We speculate that MARCH5 potentiates TLR7 and TLR4 signaling via a similar mechanism. Interestingly, this regulatory function was less potent for TLR4 signaling, probably due to the observation that other TRAFs could partially mediate TLR4 signaling to NF-κB activation [44]. Consistently, it appears that MARCH5 does not influence the activation of IRF3/7, since TANK displays no regulatory role toward TRAF3 [19].
Interestingly, mislocalization of MARCH5 to either the plasma membrane or the nucleus abolishes its function toward TLR7 signaling. An increasing number of sub-cellular organelles are functionally connected to the anti-microbial defense system. For example, it has been proposed that endosomes and lysosomes contain TLR3/7/8/9 and probably TLR4 [45]. Recognition of PAMPs by TLRs actually takes place inside these intracellular membrane structures, instead of on the plasma membrane. In addition, the endoplasmic reticulum (ER) plays an active role during the transport of TLRs to their appropriate locations. Unexpectedly, mitochondria have recently been uncovered as a new platform for sensing intracellular virus infections. Notably, the existence of cross-talk between TLR signaling and mitochondrial proteins remained unknown. Arguably, our current study reveals the first mitochondrial protein to positively regulate TLR signaling. Because mitochondria and the ER are physically connected, we expect that future investigations will uncover more intricate cross-talk between them during TLR signaling.
MARCH family proteins contain a RING finger domain and some trans-membrane motifs. Notably, many of the 11 mammalian MARCH proteins have been implicated in modulating immune functions, either directly or indirectly. MARCH8 (also known as c-MIR, cellular modifier of immune response) was demonstrated to specifically catalyze B7.2 ubiquitination and its subsequent lysosomal degradation. In addition, both MARCH8 and MARCH1 negatively modulate the expression of CD95 (Fas), TfR (transferrin receptor) and MHC class II [28]. MARCH4 and MARCH9 influence antigen presentation by MHC class I molecules. Furthermore, ectopic-expression of MARCH9 leads to the down-regulation of surface ICAM1, a co-stimulatory molecule for T and B cells [28]. It will be intriguing to examine whether other MARCH family proteins play a critical role in immune regulation.






