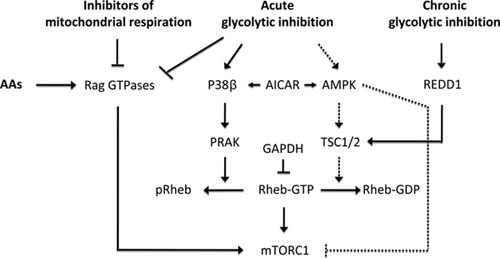
Throughout evolution, cells have developed sophisticated signaling mechanisms to balance the production and expenditure of energy to maintain energy homeostasis. During an energy crisis, cells suppress energy consuming anabolic processes and up-regulate basic catabolic routes to maintain the energy currency of the cell, Adenosine Triphosphate (ATP). The main paths of ATP generation are through glycolysis in the cytoplasm and oxidative phosphorylation in mitochondria. A major consumer of cellular energy is the mammalian Target of Rapamycin (mTOR) signaling pathway involved in the synthesis of proteins, ribosomes and lipids. mTOR exists in two structurally and functionally distinct protein complexes, mTORC1 and mTORC2, both are activated by growth factors and hormones, but only mTORC1 responds positively to nutrients and energy. Initial studies using inhibitors of glycolysis or mitochondrial respiration revealed that mTOR pathway responds to either energy source 1. A drop in ATP production leads to the immediate inhibition of mTORC1 signaling within minutes, defined as the acute response, followed by a phase of recovery and then a second long term phase of inhibition, initiated within hours and termed the chronic response 2. Inhibition of mTORC1 signaling through the chronic response is mediated by the transcriptional up-regulation of Regulated in Development and DNA Damage response 1 (REDD1) 3. In contrast, the widely accepted view is that the acute response is mediated by the AMP-Dependent Protein Kinase (AMPK), either through the phosphorylation and activation of the Tuberous Sclerosis Complex 1/2 (TSC1/2) 4, 5 or through the phosphorylation and inhibition of the mTORC1 component raptor 6 (Figure 1). However, recent loss of function studies demonstrated that acute mTORC1 inhibition by energy deprivation, can be regulated independently of AMPK through the Rag GTPases 2 (Figure 1). Likewise, Lee et al. have reported that independent of AMPK, glucose deprivation leads to Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) binding of Rheb and inhibition of mTORC1 signaling 7 (Figure 1).
能量供应紧张mTORC1调控的信号通路
In a recent study, Zheng and colleagues have brought a new player into the field of energy balance, the p38 pathway 8. The p38 pathway belongs to a MAPK signaling module, a highly conserved pathway known for its role in stress responses, including those to pro-inflammatory cytokines and UV irradiation as well as heat and osmotic shock 9. There is a body of evidence showing crosstalk between the p38 and the mTORC1 pathways 10, 11. The role of the p38 pathway in global stress responses prompted Zheng and colleagues to investigate whether p38 regulates mTORC1, following an energy deprivation episode. Initial analysis employing p38 knockout Mouse Embryonic Fibroblasts (MEFs) revealed that p38β was as an essential negative regulator of mTORC1 during energy deprivation crisis induced by 2-Deoxy-D-Glucose (2-DG), a glycolytic inhibitor. The authors then demonstrated that these effects were mediated by p38β-activated kinase PRAK. Similar to 2-DG, mTORC1 inhibition by glucose deprivation was also found to be dependent on the p38β–PRAK cascade. The authors further demonstrated that ectopic expression of PRAK inhibited mTORC1 in the absence of TSC2, whereas ectopic expression of TSC2 inhibited mTORC1 in the absence of PRAK, indicating that PRAK and TSC2 operate in parallel pathways to inhibit mTORC1. However, it is important to note that 2-DG-induced mTORC1 inhibition is impaired in TSC2 knockout cells despite normal activation of the p38β–PRAK cascade. Conversely, 2-DG activation of AMPK was unable to inhibit mTORC1 in PRAK knockout cells. This would suggest that upon 2-DG treatment, p38β–PRAK pathway and AMPK-TSC1/2 pathway might operate on a common downstream effector to inhibit mTORC1. Interestingly, 5-Aminoimidazole-4-Carboxamide 1-β-D-Ribofuranoside (AICAR), an AMPK activator triggers the p38β–PRAK cascade in the absence of AMPK and TSC2. However, it was not demonstrated whether PRAK activation led to mTORC1 inhibition in these settings. The absence of such evidence leaves the question unresolved as to whether p38β–PRAK cascade is dependent on AMPK. This is a critical issue as others have shown that AICAR, but not glycolytic or mitochondrial respiratory inhibitors, suppresses mTORC1 signaling in an AMPK-dependent manner 2.
How does PRAK inhibit mTORC1 signaling? Earlier studies showed that fusion of raptor with the c-terminal 20 amino acid portion of Ras Homolog Enriched in Brain (Rheb), the small GTPase critical for mTORC1 activation, leads to lysosomal localization of mTORC1 and its constitutive activation 12. Zheng et al. found that ectopic expression of PRAK inhibited mTORC1 activation by the raptor-Rheb variant as well as that induced by ectopic expression of wild-type Rheb, raising the possibility that PRAK acts directly on Rheb. Immunoprecipitation and kinase assays showed that PRAK can bind and phosphorylate Rheb, with the sites of phosphorylation identified as T44 and S130. Mutational analysis revealed that S130 was the critical site by which PRAK negatively regulated Rheb function, and following 2-DG treatment, this site was shown to be phosphorylated in cultured cells in a PRAK-dependent manner. Importantly, overexpression of a RhebS130A mutant prevented 2-DG-induced mTORC1 inhibition and ectopic expression of PRAK could not suppress mTORC1 activation by this mutant. Mechanistically, these findings were consistent with in vitro guanine nucleotide binding assays showing that Rheb phosphorylation results in decreased guanine nucleotide binding. Further in vitro experiments showed that S130 phosphorylation caused GTP to be more readily released from Rheb, and consistent with this, studies in cultured cells demonstrated a 2-fold reduction in nucleotide binding of Rheb in wild-type MEFs upon 2-DG treatment, as opposed to no effect in PRAK-deficient cells. This represents a novel mode of regulation of Rheb function, since only changes in GTP hydrolysis 13 or its sequestration by other proteins have been documented 7. Since the nucleotide binding capacity of the Rheb mutants was distinct, it was not possible to accurately evaluate the effect of phosphorylation on GTP hydrolysis 8. An intriguing observation made by the authors is the dependency of glycolytic inhibitors, but not mitochondrial respiratory inhibitors, on PRAK. As noted earlier, inhibition of either pathway is anticipated to decrease intracellular ATP pools and therefore a pathway that is responding to changes in ATP levels would be expected to play a role in both settings to inhibit mTORC1 signaling. Given this reasoning, it will be of interest to know whether mitochondria respiratory inhibitors also activate p38β–PRAK cascade and increase Rheb phosphorylation. It should also be noted that the regulation of mTORC1 signaling by energy-depleting agents, particularly metformin and phenformin, was shown to be dependent on the Rag GTPases 2. In the light of the results obtained by Zheng et al., it will be important to determine the role of p38β–PRAK cascade in relation to that of the Rag GTPases and whether these two pathways are integrated with one another or whether one pathway is uniquely used depending on the type of energy stress.
A number of issues remain unanswered in the field, which will prompt future studies. For instance, Inoki et al. demonstrated earlier that high concentrations of 2-DG caused osmotic shock, which led to the down-regulation of mTORC1 signaling in a TSC2-independent manner 4. Zheng et al. reported now that high concentrations of sorbitol did not require PRAK protein to down-regulate mTORC1 signaling 8; arguing that either pathway may substitute for the other or that osmotic shock alters mTORC1 signaling through yet another pathway whose identity is unknown. Likewise, Guan and colleagues reported earlier that p38-MK2, unlike p38β–PRAK cassette 8, positively regulates mTORC1 signaling by phosphorylating TSC2 10. It will be of interest to elucidate upstream events by which these distinct and opposing roles of p38 are involved in regulating mTORC1 function. Finally, it was reported that mTORC1 inhibition by either glycolytic or mitochondria respiratory inhibitors is transient and that the activity of mTORC1 recovers over time due to compensation of the alternative energy source 2. It is surprising that in this new study such a transient effect on mTORC1 signaling upon 2-DG treatment was not observed 8.
The discovery by Zheng et al. adds yet another layer of regulation of mTORC1 signaling by energy levels that highlights the importance of the cell's need to maintain tight energy homeostasis by strictly fine tuning major energy consuming processes. This study represents a third instance of a mechanism regulating mTORC1 by energy levels that argues to function independently of AMPK, the cell's master energy gauge 2, 7, 8. Finally, energy-depleting agents such as 2-DG and metformin are the object of clinical trials to evaluate their therapeutic potential for the treatment of cancers since many tumors present a hallmark for being highly dependent on glycolysis and energy levels on the whole. Therefore, a deeper understanding of the different facets of regulation of mTORC1 by energy levels will be very valuable.






