摘要:蛋白质折叠可不像叠衣服那样简单,事实上,这是生物化学中的核心问题之一,如何在有机体中由一个长长的、卷曲的氨基酸链折叠而成更复杂的三维结构,这种折叠是一个持续并且普遍的过程。了解了蛋白质的折叠,以及他们更倾向于形成的结构,研究人员才能在研究其功能方面更进一步。MIT和McGill的研究人员一起开发出了一种新的算法,在一台笔记本电脑上就可以确定某种蛋白质的基本化学性质,之后可以扫描出该蛋白可能的几种结构形态。
研究蛋白质折叠的重要性在于错误的折叠可能会导致各种疾病的发生,比如阿兹海默症、帕金森氏病、顿氏舞蹈症、肺气肿以及囊胞性纤维症等。研发更好的蛋白质折叠建模技术对于创造更有效的药物来治疗这类及其他相关疾病来说是至关重要的。
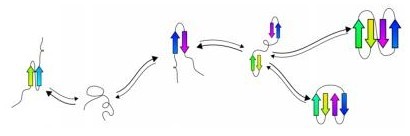
Computational methods of modelling protein folding have existed for a couple of decades. But what McGill researcher Jérôme Waldispühl of the McGill Centre for Bioinformatics has done, working with collaborators from MIT, is to develop algorithms that can work from a laptop computer to examine a protein's fundamental chemical properties and then scan a number of possible protein shapes before predicting the final form that the protein is likely to take.
The results have been impressive. Whereas classical techniques for predicting protein folding pathways required hundreds of thousands of CPU hours to compute the folding dynamics of 40 amino acids proteins, the program tFolder implemented by Solomon Shenker – a former McGill under-graduate student now at Cornell – has been able to predict correctly in 10 minutes on a single laptop, a coarse-grained representation of the folding pathways of a protein with 60 amino acids.
Waldispühl and his students continue to work on their algorithm to improve its success rate at predicting protein folding with broader categories of proteins including some that are important in DNA-binding. The research was recently presented at the 15th Annual International Conference in Research in Computational Molecular Biology (RECOMB 2011).
https://esciencenews.com/articles/2011/06/07/protein.folding.made.easy






