本文转载自“学术头条”微信公众号。
1957 年,John C. Kendrew 和 Max F. Perutz 通过 X 射线晶体学确定了第一个蛋白质结构。不久之后,Christian B. Anfinsen Jr. 提出蛋白质的结构在热力学上是稳定的,似乎可以根据蛋白质的氨基酸序列来预测蛋白质的三维结构。
然而,蛋白质的结构复杂性远超人们的想象。依据中心法则,蛋白质主要是由 DNA 转录成 RNA,再翻译成肽链后组装而来,一个蛋白质分子是由一条或几条多肽链组成,多肽链则折叠成特有的形状。同时,蛋白质分子的专一形状是由4个层次的结构决定的,包括一级、二级、三级和四级结构,前一级结构决定后一级结构。
其中多肽链的氨基酸序列是一级结构,一级结构中的部分肽链卷曲或折叠产生二级结构。二级结构经过一系列的构象改变形成三维结构即三级结构,一般为球状或纤维状。三级结构有特定的结构域,形成结合位点或调节位点,可以结合特定结构的物质,行使特定的功能。两条或两条以上的多肽链组成的蛋白质,可以形成四级结构。

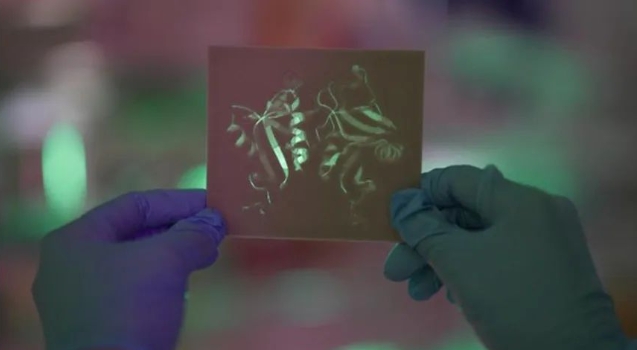
图 | 蛋白质 3D 结构(来源:Nat Commun)
而近年来,随着冷冻电子显微镜技术的发展,可以在没有结晶样本的条件下观察蛋白质结构,使得蛋白质结构研究有所进展。不过,冷冻电镜是非常昂贵的设备,只有极少数的实验室才有条件配备,对于广大科研工作者非常不友好。因此,生命科学界亟需新的方法解决蛋白质折叠问题。
在过去 25 年中,国际蛋白质结构预测大赛(CASP)一直关注这个领域的进展,试图寻找能够完美解决蛋白质折叠问题的计算机模型。直到第 14 届大赛 CASP14 大赛成功举办,DeepMind 旗下的 AlphaFold 系统在蛋白质结构预测方面表现出了无与伦比的准确性。
该比赛的评价方式是将参赛者提供的解决方案与“黄金试验标准”进行对比,用 GDT 评分衡量准确性,范围为 0-100,GDT 分数在 90 分左右,即可视为对人类实验方法具备竞争力。而 DeepMind 旗下的 AlphaFold 系统总分竟然达到了 92.4,和实验的误差在 1.6,即使是在最难的没有同源模板的蛋白质上面,这个分数也达到了了恐怖的 87.0 。
同时,AlphaFold 的神经网络能在几分钟内预测出一个典型蛋白质的结构,还能预测较大蛋白质(比如一个含有 2180 个氨基酸、无同源结构的蛋白质)的结构。该模型能根据每个氨基酸对其预测可靠性进行精确预估,方便研究人员使用其预测结果。

图 | 研究人员使用 RoseTTAFold 预测的人类白细胞介素 12 与其受体结合的 3D 视图(来源:UW Medicine Institute for Protein Design)
不仅如此,RoseTTAFold 所需的计算耗能与计算时间均比 AlphaFold2 还要低:仅用一台游戏计算机,在短短十分钟内就可以可靠地计算出蛋白质结构。更值得注意的是,RoseTTAFold 的代码和服务器完全免费提供给科学界!
同样在今年 7 月份,DeepMind 创始人兼首席执行官 Demis Hassabis 也在 Nature 杂志上分享了AlphaFold的开源代码,并发表了系统的完整方法论,详尽细致说明 AlphaFold 是如何做到精确预测蛋白质3D结构的。也就是说,这款强大蛋白质结构预测模型已经是完全免费的。
至此,两种强大的基于人工智能的蛋白质结构预测模型全部免费开放,科研工作者可以随时利用这两款模型获取蛋白质的空间结构,而无需对蛋白质进行结晶或使用昂贵的冷冻电镜进行研究。
在同步配发的评论文章里,Science 杂志的主编 Holden Thorp 对此表示,“首先,它解决了困扰生命科学近 50 年的蛋白质折叠问题,好比物理学中的引力波,科学家们数十年如一日,坚持不懈最终才攻克这一难题;其次,这一技术改变了未来结构生物学的规则,就像冷冻电镜那样,加速生命科学的发展;此外,完全免费意味着它是真正适合所有人的蛋白质预测模型。”
关键词:
Science