囊性纤维化是白人中最常见的影响生存寿命的遗传性疾病,然而,对它的治疗却一直没能取得太大的进展。最近,Bonnie W. Ramsey和Jane Davies等研究了ivacaftor治疗囊性纤维化的效果,并将结果发表在了本期的NEJM文章《A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation》中。
囊性纤维化(cystic fibrosis,CF)是一种累及外分泌的遗传病,主要以慢性梗阻性肺部病变、胰腺外分泌功能不足和汗液电解质异常升高为主要特征。CF是由于编码囊性纤维化跨膜转录调节因子(cystic fibrosis transmembrane conductance regulator, CFTR)蛋白的第7号常染色体基因突变导致的。CFTR是一种氯离子通道蛋白,在呼吸道表面,它主要表达于黏液-纤毛清除系统的黏液腺体中,在黏液纤毛清除和自我防御方面起着重要的作用。
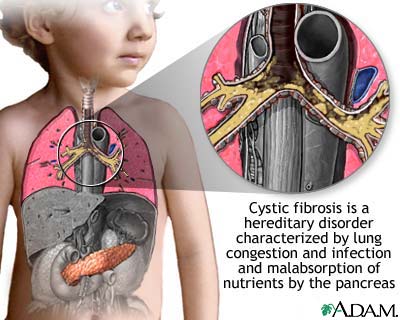
CF突变后使气管上皮细胞对氯离子的渗透性大大降低,而对钠离子的渗透性却大大提高,这就导致了黏液腺体的阻塞,使气管腔中的黏液性分泌物大大减少,气管的“清道夫”——黏液纤毛不能发挥正常作用,就会导致慢性细菌感染和气管炎症,最终的结果便是肺功能的不可逆损伤。
最近,科学家们将目光投向了囊性纤维化的缺陷基因——囊性纤维化跨膜转导调节蛋白(cystic fibrosis transmembrane conductance regulator, CFTR)基因,CFTR的缺陷在此病的发生过程中起到了很大的作用,而ivacaftor则正是通过提高CFTR的作用来达到治疗目的的,这种口服药可以增加细胞表面CFTR通道的开放时间,体外实验证明,ivacaftor可以提高G551D—CFTR蛋白的氯离子转运活性,从而在源头上阻断CF的发病。
这项随机双盲,安慰剂控制试验的被试均为12岁以上的囊性纤维化患者,他们都至少有一个G551D-CFTR基因突变。试验共历时48周,其中试验组的84名被试接受每12小时150mg的ivacaftor治疗,对照组则接受安慰剂治疗。
试验的结果还是非常鼓舞人心的。试验24周以后,试验组的预计一秒钟用力呼气量(forced expiratory volume in 1 second,FEV1)相对于基线水平比对照组增加了10.6%。ivacaftor对肺功能的改善同样在用药后两周便显现了出来。试验组的肺部病变恶化率较对照组下降了55%,试验结束时,实验组的平均体重比对照组多增加了2.7kg(3.1kg vs. 0.4kg)。对于汗液电解质水平的改善情况,ivacaftor也要明显的优于安慰剂,在试样24周时,试验组的汗液氯离子浓度下降了48.7mmol/L,而对照组则只下降了0.8mmol/L。(对于以上试验结果,p<0.001)。同时,试验组和对照组副作用的发生率相似,严重副作用的发生率则比对照组低很多。(24% vs.42%)
试验结果说明ivacaftor对于囊性纤维化的患者可以很好的改善其临床症状,同时可以降低肺部疾病恶化的风险。
那么,研究者的这个试验结果是否意味着人类已经彻底战胜了囊性纤维化这一令人闻之色变的遗传病呢?答案却仍然不甚乐观。
CF的传统治疗主要是以恢复黏液纤毛排送系统功能和抗感染为主,既然CFTR的突变是通过影响黏液的清除过程也引起的CF,那么医疗工作者便很自然的将目光聚焦在了提高纤毛黏液的清除上,硫酸类肝素和人类重组DNA酶便是这类药物的代表。抗炎在CF的治疗中也发挥着重要的作用。在CF的致病菌中,最严重的是假单胞菌的感染。研究表明,感染假单胞菌患者口服环丙沙星,或者吸入黏菌素也可以起到一定的消炎作用,从而控制疾病的进展。
那么目前最具前景的Ivacaftor是否真的能够使患者肺功能的恶化情况停止甚至逆转还是需要时间来评判的。由于囊性纤维化患者肺部病变的进展太过于缓慢,对被试接下来几年的随访仍然是必不可少的。
其次,研究者需要解决的另一个问题是ivacaftor能否激活细胞表面CFTR的其他等位基因,因为G551D基因的突变在患者中毕竟只占4~5%,一旦如此,必将有更多的患者从中受益。
第三,婴幼儿和儿童使用ivacaftor以及长期使用这一药物是否安全都是亟待解决的问题。从囊性纤维化的发病过程来看,尽管胰腺等很多系统的损害早在胚胎时期就已经形成了,但最严重的并发症——肺部和肝脏的病变——却是在出生以后才开始的。那么一个很现实的问题就摆在了研究者面前:能够预防性使用ivacaftor以防止囊性纤维化患者肺部病变的发生?在美国的大多数州,囊性纤维化已经被包含在新生儿筛查的范围中。如果拥有G551D-CFTR基因突变的患者均能在肺病病变发展前得到诊断,同时在及时予以ivacaftor治疗后能够显著的改善生活质量的话,我们也许就真的可以宣告囊性纤维化时代的终结了。
相关英文论文摘要:
A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation
BACKGROUND Increasing the activity of defective cystic fibrosis transmembrane conductance regulator (CFTR) protein is a potential treatment for cystic fibrosis.
METHODS We conducted a randomized, double-blind, placebo-controlled trial to evaluate ivacaftor (VX-770), a CFTR potentiator, in subjects 12 years of age or older with cystic fibrosis and at least one G551D-CFTR mutation. Subjects were randomly assigned to receive 150 mg of ivacaftor every 12 hours (84 subjects, of whom 83 received at least one dose) or placebo (83, of whom 78 received at least one dose) for 48 weeks. The primary end point was the estimated mean change from baseline through week 24 in the percent of predicted forced expiratory volume in 1 second (FEV1).
RESULTS The change from baseline through week 24 in the percent of predicted FEV1 was greater by 10.6 percentage points in the ivacaftor group than in the placebo group (P<0.001). Effects on pulmonary function were noted by 2 weeks, and a significant treatment effect was maintained through week 48. Subjects receiving ivacaftor were 55% less likely to have a pulmonary exacerbation than were patients receiving placebo, through week 48 (P<0.001). In addition, through week 48, subjects in the ivacaftor group scored 8.6 points higher than did subjects in the placebo group on the respiratory-symptoms domain of the Cystic Fibrosis Questionnaire–revised instrument (a 100-point scale, with higher numbers indicating a lower effect of symptoms on the patient's quality of life) (P<0.001). By 48 weeks, patients treated with ivacaftor had gained, on average, 2.7 kg more weight than had patients receiving placebo (P<0.001). The change from baseline through week 48 in the concentration of sweat chloride, a measure of CFTR activity, with ivacaftor as compared with placebo was −48.1 mmol per liter (P<0.001). The incidence of adverse events was similar with ivacaftor and placebo, with a lower proportion of serious adverse events with ivacaftor than with placebo (24% vs. 42%).
CONCLUSIONS Ivacaftor was associated with improvements in lung function at 2 weeks that were sustained through 48 weeks. Substantial improvements were also observed in the risk of pulmonary exacerbations, patient-reported respiratory symptoms, weight, and concentration of sweat chloride. (Funded by Vertex Pharmaceuticals and others; VX08-770-102 ClinicalTrials.gov number, NCT00909532.)
英文论文链接:https://www.nejm.org/doi/full/10.1056/NEJMoa1105185?query=featured_home






