
几十年来,新药的研发就像是一场试验与误差之间的比赛,数以百万计的候选药物最终只有极少数被成功研发。最近,研究人员发明了一种计算机算法,为今后的药物研发带来了一个良好的开端。通过计算机分析药物的化学结构,研究人员可以看到它是否能够与某个生物学靶标(例如蛋白质)相结合或对接。这样就可以发现因药物与某些与生物靶标结构相似蛋白的非预期结合而引发的药物毒副作用。
上周,研究人员提出了一个工作计划,以大量预测公共数据库里的药物和蛋白质潜在的对接可能。纽约大学Langone医学中心的药理学家Timothy Cardozo曾经出席了11月19日由美国国家卫生研究院(National Institutes of Health)在马里兰州举办的“高风险高回报”研讨会,对于这个项目,他评价说:“这是有史以来人们做的最大的药物对接计算”。因此,一个叫做Drugable的网站(drugable.com)应运而生,目前仍然在测试阶段。它由美国国立图书馆(NLM)支持,计划在未来免费开放。通过这个网站,药物研究人员能够在药物化学结构的基础上预测新药如何在人体内发挥作用以及在何处发挥作用。
Cardozo承认,计算机模拟在药物开发过程中只是一个初始步骤。在预测某个蛋白是否能与化合物结合后,药物开发者必须在细胞中测试药物在相同蛋白上发挥的实际功效,以及在不同情况下药物的用量。随后再进行动物试验,如果幸运通过动物实验,接下来就是人体试验。但是加州大学旧金山分校的计算生物学家Brian Shoichet指出,这些后续的数据往往仅由制药公司持有,一些公共的数据库如PubChem,尽管有药物在酵母细胞中的试验数据,但这些数据也有可能是错误的。
尽管如此,科学家们已经证实,该算法可以为药物研发提供一些捷径。在2012年,Shoichet和研究人员在剑桥的诺华生物医学研究所开发了一种算法,该算法可以在药物化学结构相似性的基础上预测药物的副作用。当研究人员对656批药品和73种生物靶标的相互作用进行测试时,他们发现采用该算法预测出了几百种以前未知的相互作用——并且这些被测试出的副作用有一半是真实存在的。而对于已知的药物,Shoichet说,这种类型的计算可以提供一种快速的方法来确认药物与蛋白靶标的相互作用。
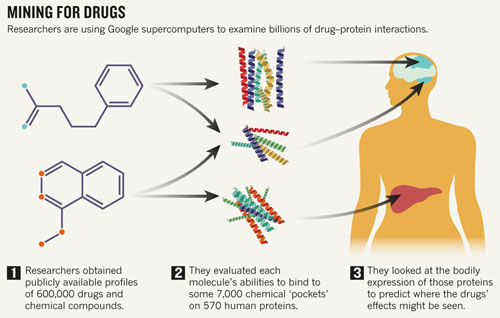
要预测那些未知化合物与靶标蛋白如何在体内相互作用更具有挑战性。在建立网站的时候,Cardozo的团队从PubChem和欧洲生物信息研究所的ChEMBL数据库搜集了约600000种分子,这些分子又可以组合成数百万的化合物。这个研究团队评估了这些分子与7000种人类蛋白结合口袋结合的程度,并将这些数据也都纳入到数据库中。计算巨头谷歌为研究人员提供了相当于超过1亿小时的计算机处理器,帮助他们完成这项庞大的工作。
该团队又提出了通过对接分数为大约40亿潜在的药物-蛋白互作程度进行排名。随后,该团队又汇总了已有靶标蛋白与NLM蛋白表达的综合数据,显示不同基因所编码的蛋白在人体的表达部位。这就使得研发人员可以预知药物在人体内的下一个走向,Cardozo指出,如果某种成药被认定为在某个组织的蛋白质中高水平表达,那么这说明该药物很可能会对这个组织发挥作用。
诺华研究院的研究员Jeremy Jenkins表示,多年来,制药公司一直在做类似的预测。他还说,尽管拥有1.5亿个公共和专有化合物,诺华公司从来没有在药物与靶标分析时有Drugable一般的效率。
Cardozo希望Drugable能在评估精神类药物时提供特殊的帮助,因为此类药物的作用方式通常难以衡量。作为示范,Cardozo的研究小组为氯氮平和氯丙嗪进行了靶标互作计算,这两种药常被用来治疗精神分裂症。
正如预期的那样,计算结果表明两种药物与神经递质血清素和多巴胺受体结合最为强烈,这两种受体在进行高级信息处理的大脑部位表达。但是也能用来治疗抑郁症的氯氮平,还可以强烈绑定到一种名为DRD4的多巴胺受体,该受体在大脑中负责情绪调节部位——松果体中表达。
该小组还发现,氯氮平也能与大脑中调节唾液分泌的受体结合,因此唾液分泌过多也是氯氮平已知的一种副作用。虽然这种药物调节情绪和唾液分泌的的生化解释在之前就已经提出,但是Cardozo认为Drugable才是用来揭示药物作用机制最合理的渠道。






