本文转载自“药时代”。
为了成功开发和制造仿制药品,申请人应期望其产品与参比制剂(RLD):在相同的使用条件下,在药学上一致,即具有相同的活性成分、剂型、规格和给药途径 ;与参比制剂具有生物等效性,即在活性药物成分的吸收速率和程度上没有显着差异;因而,疗效上相当,即可替代参比制剂,即仿制药与相对应的参比制剂具有相同的安全性和功效。
根据21 CFR 320.24,可以使用不同类型的证据来建立药学上等同的药品直接的的生物等效性,包括体内测试或体外测试,或两者都有。用于证明生物等效性的方法的选择取决于研究的目的、可用的分析方法和药物产品的性质。根据这一规定,申请者必须使用21 CFR 320.24中规定的最准确、灵敏和可重现的方法进行生物等效性测试。作为选择仿制药产品开发方法的初步步骤,申请人可参考以下指导原则草案:根据简化新药申请(ANDA)提交的药物动力学终点的生物等效性研究工业指导原则草案(2013年12月)。
为了进一步促进仿制药产品的可及性和协助仿制药行业确定最适合开发药物的方法并产生支持ANDA批准所需的证据,FDA发布特定产品指南,描述FDA目前对如何开发与特定参比制剂临床等效的仿制药的思考和期望。
最新发布的指导原则82个(新增 21 个、修订版 13个、最终版 48个)如下。
新增指导原则草案 21 个
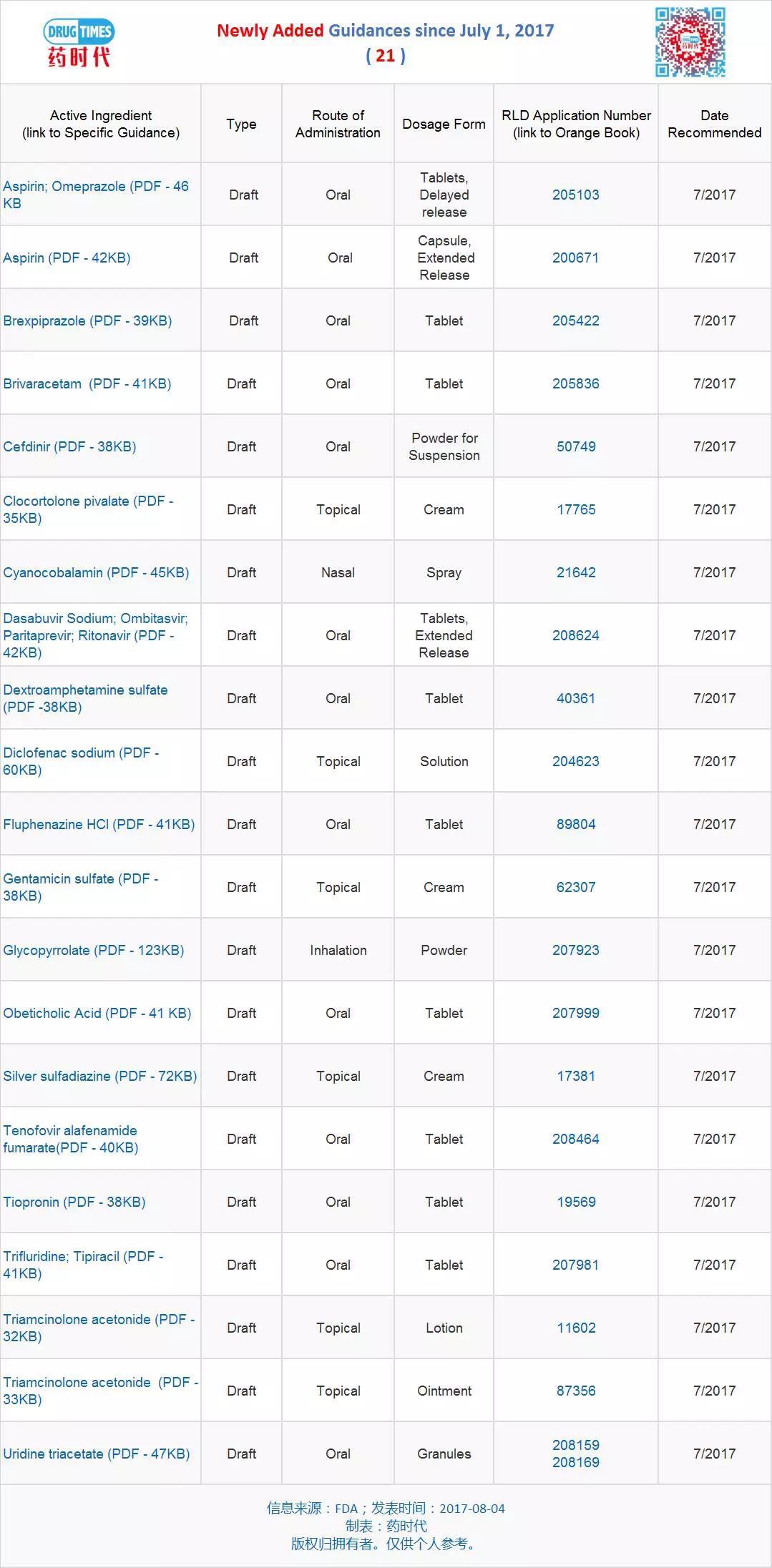
新修订指导原则草案 13 个
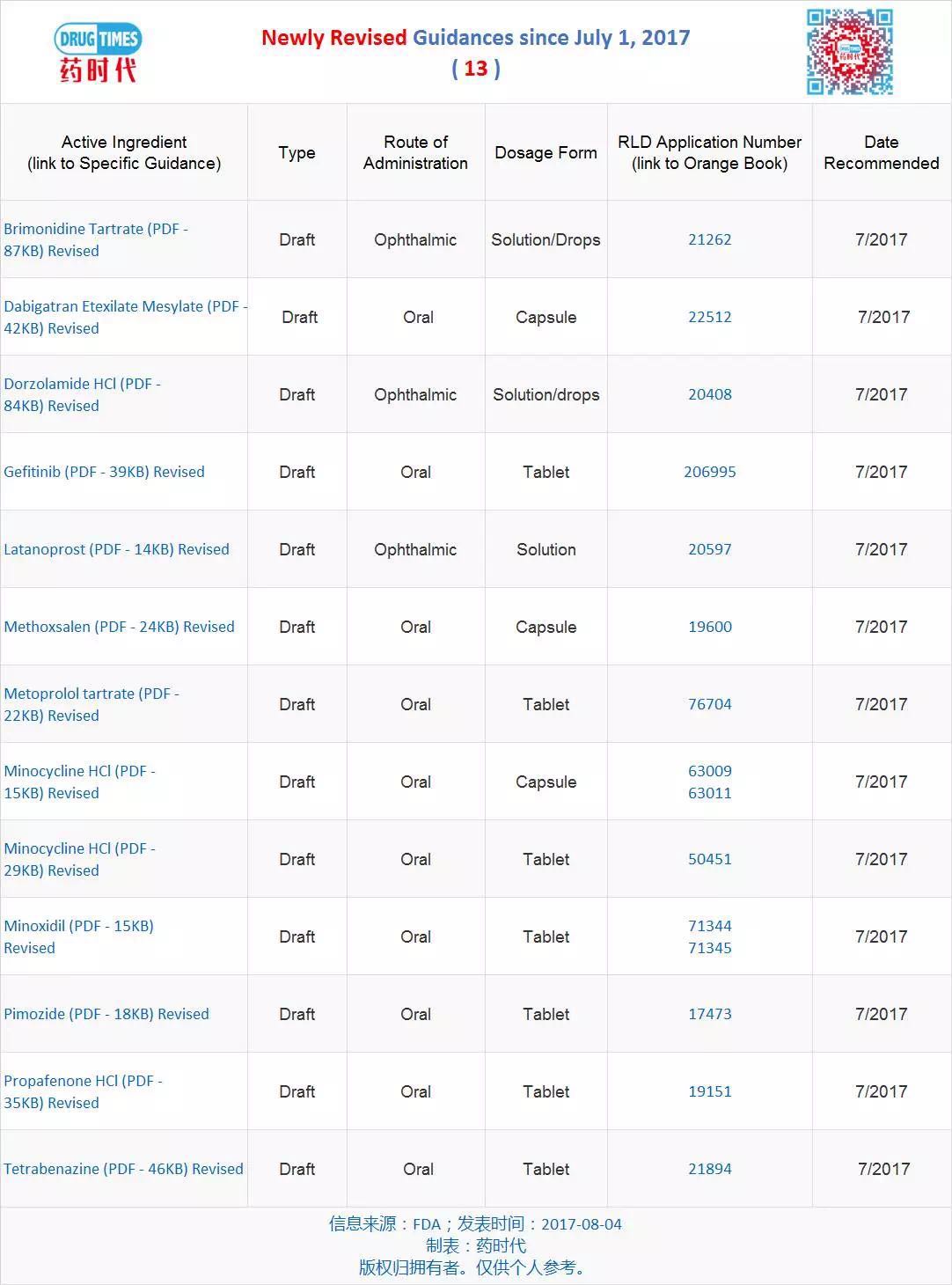
新增最终指导原则 48 个