作者:Joice HU,缔脉医学副总监
由于恶性肿瘤的高发病率和死亡率,抗肿瘤药物的研发已然成为目前以及未来整个新药研发中最重要的部分。经过几十年的不懈努力,我国抗肿瘤药物的临床研究已经取得了显著进展,积累了一定的经验,由此带来了肿瘤内科的快速发展、肿瘤内科和肿瘤综合治疗水平的显著提高。但是近年来,随着肿瘤免疫,精准治疗等新治疗的不断涌现,以及政策法规对与临床研究的相应更新,对临床研究相关人员的要求也在不断提高。基于此巨大的需求,DIA聚集临床和工业界的多方专业资源,开展了OnSITE(Oncology Study Investigator Training/Engagement肿瘤研究者培训)项目,聚焦肿瘤药物研发相关政策法规,GCP和伦理审核,肿瘤临床研究设计,疗效和安全性评估,风险管控和真实世界研究8大模块,于11月16-17日,面向临床研究者和相关专业人员,开展了首期2天的旗舰培训课程。
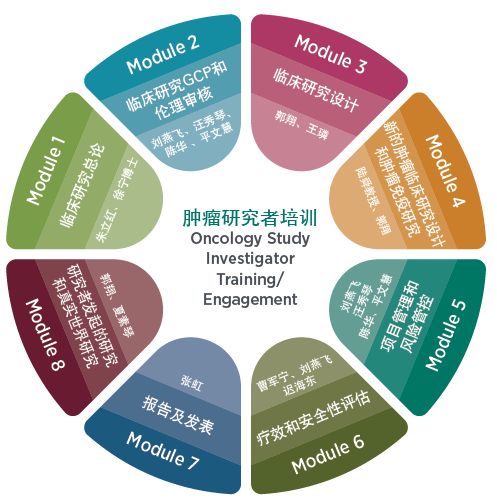
OnSITE八大模块
第一部分:肿瘤临床研究相关法规介绍
讲者:汪旭
武田制药高级注册事务经理

在过去,由于中国药品注册申请时限长,制约了自主创新和跨国研发,也延迟了仿制药的上市,最终影响了药物的可及性。自2015年来,一系列的政策法规相继出台,在提高审评质量的同时,也通过优先审评制度、沟通交流制度、临床试验默认许可制度等一系列法律法规简化审批程序、鼓励研究创新。
在此一系列的改革之下,目前根据CDE网站公示信息所示,通过默示许可制获得临床试验批准的时限已显著的缩短,最短的案例仅为2个月。而对于临床急需药品,根据事先的沟通交流确定,可基于可合理预测临床获益的替代终点批准,也可以基于早期中期数据显示的较现有治疗的明显优势,在要求上市后确证性临床试验的基础上提前批准。而对于罕见疾病,在境外批准的基础上证明无种族差异后也可在技术审评后获批。
政策的改革带来了机遇,使我国新药研发时间与欧美的差距缩短,也有利于肿瘤药物的研发和准入环境。同时,也带来了挑战,创新研发的进步还需要各相关方相互配合共同完成。
第二部分:临床研究伦理审核
新形势下肿瘤疾病
创新药物研究者的伦理要求
讲者:陆麒
上海仁济医院伦理办公室主任
上海市医学伦理学会副秘书长

递交临床研究的伦理申请,需要首先明确伦理审查的具体要求,包括研究方案、知情同意书、招募材料、研究者资质和其他的相关材料,以及这个材料的要求。临床研究在科学性的基础上,需要明确试验的风险和获益,确保受试者知情同意的完全告知、充分理解和自主选择, 并注意受试者的医疗和保护、隐私和保密。
不同的临床研究在伦理审查时也有不同的关注点。比如观察性研究,要警惕隐藏干预,注意敏感信息保护,知情同意的获得;而四期临床研究要注意药物的适应症和潜在的市场推销。在未来,随着基因治疗、干细胞治疗的发展,这些研究的伦理审查也将面对新的挑战。
最后,陆主任总结了伦理审查的48字箴言。
“知己知彼、百战不殆
不争时间、确保质量
事必躬亲、了然于胸
精诚合作、互谅互助
加强沟通、及时答复
伦理是友、保驾护航”
第三部分: 肿瘤临床研究设计
肿瘤临床试验的背景及重要性,
试验方案中临床医学角度的考量
讲者:黄薇
百济神州医学部执行总监

随着肿瘤免疫治疗的发展,肿瘤治疗已经进入了个体化治疗的PHC2.0时代。肿瘤免疫研究相比以往传统非免疫治疗的研究,在研究设计、研究终点、人群选择及安全谱等方面都有所不同, 比如适应性设计;由于免疫治疗的延迟反应,设立OS作为联合主要终点;选择PD-L1+患者群;警惕免疫相关不良反应。
除了药物的注册临床研究,研究者发起的临床研究对促进创新研发也是一个经济有效的方式。
黄博士对目前肿瘤研究所选取得研究终点进行了详细得介绍和比较(见表1)。此外,研究人群(入排标准)的选取,医学监察对一个研究的成功也是至关重要的。
表1 抗肿瘤药物审批所用重要临床试验终点的比较▼

在安全性方面,免疫检查点药物的安全性特征和既往的治疗有很大的不同,需要加强免疫相关不良反应(irAE)的识别,早期发现和及时治疗是处理此类别不良反应的关键。
创新性临床试验的现状和未来
讲者:陆舜
上海交通大学附属胸科医院
上海市肺部肿瘤临床学中心主任

随着肿瘤药物的不断创新,肿瘤临床试验也面临着巨大的革新。相比传统化疗药物,分子靶向药物由于其毒性小,无需周期性给药,可以连续使用,给药剂量方面,最佳生物剂量的概念也代替了传统的MTD,可以固定剂量给药,无需计算体重/体表面积。
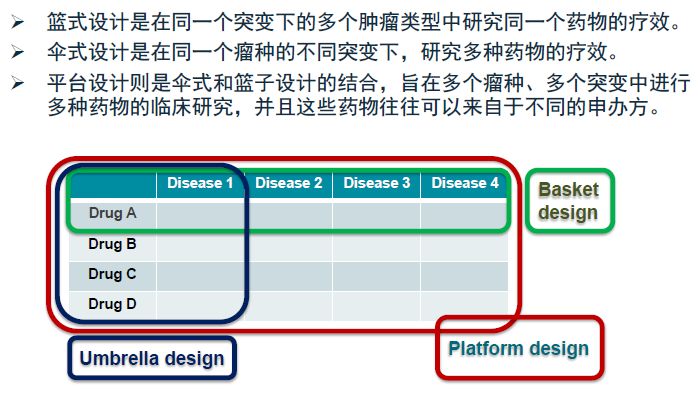
新型的抗肿瘤药物可以通过生物标志物来预测疗效,因此,这些药物在验证临床疗效的同时,需要进行伴随诊断的开发,这就是整合生物标志物的药物的研发。肿瘤临床研究设计的未来将通过伴随诊断以生物标志物来选择患者(研究样本量相对小,但筛选样本量大),对I期临床研究进行扩展,采用伞式和篮氏的研究设计,研究终点考虑PFS(无进展生存期)和RR(反应率)。而研究成功的秘诀在于找到驱动基因和目标人群,研究的速度也非常重要。
中国的创新药物研究设计需要在经典中不断推陈出新,建立在科学合理的基础上进行小修小补,例如基于现有的数据和人群特点找到快速筛选的方法;对传统理念也需要有合理的突破,比如ADJUVANT研究就跳出了RADIANT研究先做化疗再和安慰剂进行对比的传统设计,直接和化疗药物进行头对头比较。无缝设计可以帮我们更高效的确定生物标志物的界值或状态。篮氏/伞式设计和真实世界研究也可以给我们提供更多科学有效的创新方式。
试验方案中的统计学考量,
肿瘤药物临床试验设计概述,
破解临床研究结果的密码
讲者:邱婧君
百济神州生物统计副总监

试验方案中的统计学考量这一部分介绍了临床研究中的统计学基础知识,包括描述统计和统计推断中的基本概念。此外,在肿瘤研究中由于多个终点(如联合主要终点)、多此分析(如适应性设计、中期分析)、多臂/多剂量组,多亚组分析的使用,需要使用多重比较的统计学方法。通常我们接受5%的假阳性率,但每一次额外的比较都会增加至少一个假阳性发现的总几率,因此,我们需要考虑总体I类错误(FWER)并进行多重性校正。在样本量计算时,除了I类错误(体现为α值)和II类错误(体现为检验效能)外,我们还需要考虑随机比率,界值(在非劣效或者生物等效性研究中),联合终点,中期分析等多种因素,入组率、研究间期和脱落率也需要考虑,尤其是在观察生存时间的研究中。
肿瘤药物临床试验设计的部分介绍了几种目前肿瘤研究中常用的新型研究方法:适应性设计(图1)和无缝设计(图2)已逐渐广泛应用于抗肿瘤药物的研发中,以期更有效地讲优选的药物或治疗方案提供给患者;而篮氏、伞式、平台设计(图3)等也为抗肿瘤药物的研发提供了更多的可能性。
图2 无缝设计▼
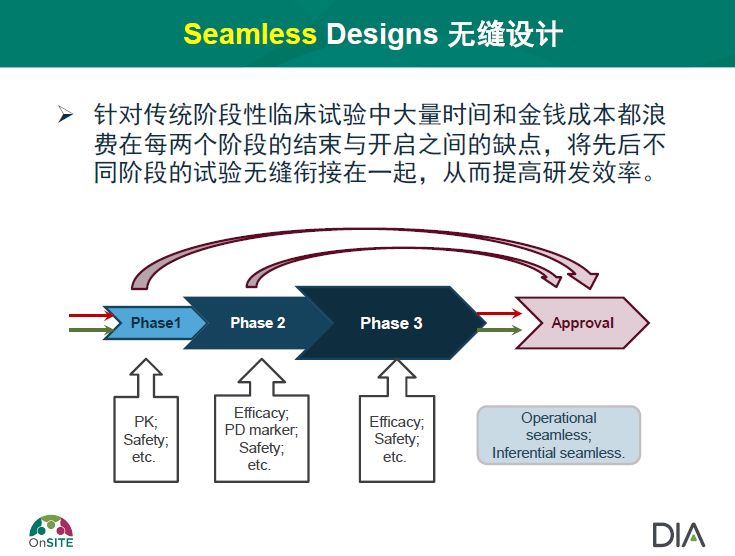
图3 篮氏、伞式、平台设计▼
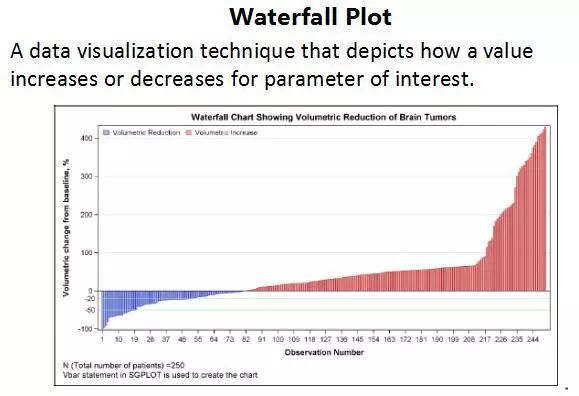
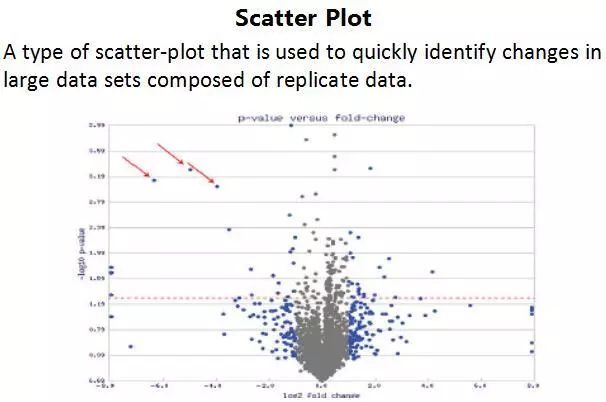
在解读临床研究的数据时,RR(相对危险度)和OR(优势比)的概念一直是容易混淆的部分。邱老师对这些概念做了详细的解释(表2),而在肿瘤药物临床研究中更常见的Hazard Ratio(风险比,当反应变量为事件发生的时间时,一个治疗组的事件的风险与比较组的风险之比)与RR相似,但是考虑了时间的因素。除了生存分析中常见的Kaplan-Meier曲线外,我们还了解了瀑布图、火山图、森林图等不同的数据表现形式,更有利于对疗效和安全性的解读(图4)。
表2 RR与OR的比较▼
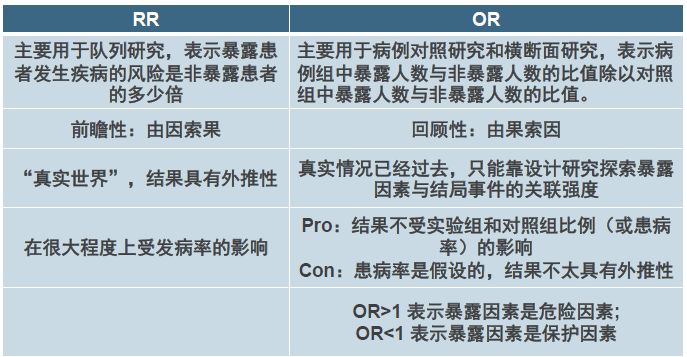
图4 生存分析中的常用图表▼